Wie gut sind Vorhersageprogramme für Proteinstörungen eigentlich?
Anzeigen


Unordnung in Proteinen ist entscheidend für die biologische Funktion, und strukturelle Störungen der Proteine sind allgegenwärtiger, als Sie vielleicht denken. Proteine mit ungeordneten Regionen können klebrig sein und innerhalb und zwischen den Zellen verklumpen. Sie sind direkt an einer Reihe von neurodegenerativen Erkrankungen beteiligt. Daher ist es sehr wichtig, ungeordnete Regionen in Proteinen identifizieren zu können.
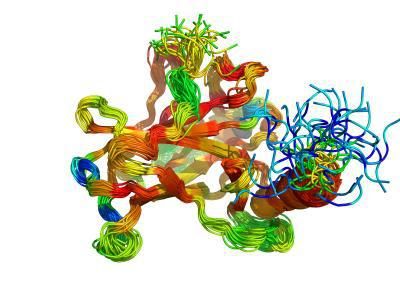
Die AU-Forscher Jakob T. NIelsen und Frans Mulder präsentieren Analysen mit NMR-Daten als Maßstab für den Test von Programmen zur Vorhersage von Proteinstörungen. Die Abbildung zeigt die NMR-Ensemblestruktur für die Kerndomäne des Proteins p53, gefärbt nach CheZOD Z-Scores.
Dr. Jakob Toudahl and Assoc. Prof. Frans Mulder at Department of Chemistry and Interdisciplinary Nanoscience Center, Aarhus University
Leider ist es schwierig und zeitaufwendig, die strukturellen Neigungen von Polypeptiden experimentell zu charakterisieren, und deshalb sind bioinformatische Methoden zur Vorhersage von Proteinstörungen aus der Sequenz unerlässlich.
In den letzten Jahren haben viele Bioinformatiker daher Algorithmen konstruiert, um diejenigen Peptidsequenzen, die sich falten, von denen zu unterscheiden, die sich nicht falten. Diese Algorithmen können auf verschiedenen "Merkmalen" basieren, die sich aus physikalisch-chemischen Parametern (wie Ladung oder Hydrophobie einer Aminosäure) ableiten und die evolutionäre Verwandtschaft betrachten.
Nun, da viele solcher Vorhersageprogramme verfügbar geworden sind, ist es von offensichtlichem Wert, eine Art Benchmark zu haben, um die Vorhersagen zu validieren und zu testen. Um dieses Dilemma zu lösen, generierten und validierten Nielsen und Mulder einen repräsentativen experimentellen Benchmarking-Satz von ortspezifischen und kontinuierlichen Störungen unter Verwendung von hinterlegten chemischen NMR-Shift-Daten für mehr als hundert ausgewählte Proteine. Anschließend analysierten sie die Leistung von 26 weit verbreiteten Methoden zur Vorhersage von Störungen und fanden heraus, dass diese deutlich variieren.
Der in ihrer Forschung vorgestellte Vergleich soll Proteinwissenschaftlern auf der ganzen Welt helfen, besser informierte Entscheidungen darüber zu treffen, welche Programme am besten geeignet sind.
Hinweis: Dieser Artikel wurde mit einem Computersystem ohne menschlichen Eingriff übersetzt. LUMITOS bietet diese automatischen Übersetzungen an, um eine größere Bandbreite an aktuellen Nachrichten zu präsentieren. Da dieser Artikel mit automatischer Übersetzung übersetzt wurde, ist es möglich, dass er Fehler im Vokabular, in der Syntax oder in der Grammatik enthält. Den ursprünglichen Artikel in Englisch finden Sie hier.
Originalveröffentlichung



Weitere News aus dem Ressort Wissenschaft




















































