How good are protein disorder prediction programs actually?
Advertisement



Disorder in proteins is vital for biological function, and structural disorder in protein is more pervasive than you might think. Proteins with disordered regions may also be sticky, and clump together inside and between cells, and are directly implicated in a number of neurodegenerative diseases. Thus, being able to identify disordered regions in proteins is highly important.
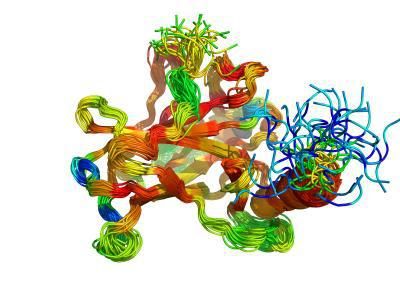
AU researchers Jakob T. NIelsen and Frans Mulder presents analysis using NMR data as benchmark for testing protein disorder prediction programs. Illustration shows NMR ensemble structure for the core domain of the protein p53 colored according to CheZOD Z-scores.
Dr. Jakob Toudahl and Assoc. Prof. Frans Mulder at Department of Chemistry and Interdisciplinary Nanoscience Center, Aarhus University
Unfortunately, it is challenging and time-consuming to characterie the structural propensities of polypeptides experimentally, and therefore bioinformatics methods for predicting protein disorder from sequence are indispensable.
Over recent years many bioinformaticians have therefore constructed algorithms to differentiate peptide sequences that will fold from those that do not, and these algorithms can be based on various 'features', derived from physicochemical parameters (like charge or hydrophobicity of an amino acid) as well as looking at evolutionary relatedness.
Now that many such prediction programs have become available, it is of obvious value to have some kind of benchmark to validate and test the predictions. To resolve this quandary, Nielsen and Mulder generated and validated a representative experimental benchmarking set of site-specific and continuous disorder, using deposited NMR chemical shift data for more than a hundred selected proteins. They then analysed the performance of 26 widely-used disorder prediction methods and found that these vary noticeably.
The thorough comparison presented in their research will help protein scientists around the globe to make better informed choices about which programmes are best to use.
Original publication




Other news from the department science



















































