Se pueden deducir tres reglas de oro para diseñar enzimas optimizadas para reacciones químicas
Instrucciones de montaje de las enzimas
Anuncios



En biología, las enzimas han evolucionado durante millones de años para impulsar reacciones químicas. Científicos del Instituto Max Planck de Dinámica y Autoorganización (MPI-DS) han deducido ahora reglas universales que permiten el diseño de novo de enzimas óptimas. Como ejemplo, consideraron la reacción enzimática de ruptura de un dímero en dos moléculas monoméricas. Teniendo en cuenta la geometría de este complejo enzima-sustrato, identificaron tres reglas de oro que deben tenerse en cuenta para construir una enzima funcional.
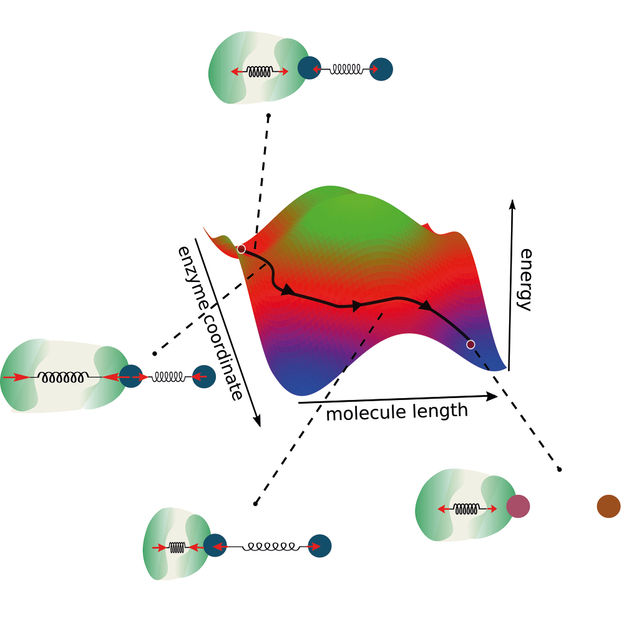
Un nuevo modelo de reacciones enzimáticas amplía la coordenada de reacción tradicional al considerar el acoplamiento entre la enzima y la molécula. De este modo, se abren vías alternativas para las reacciones enzimáticas, lo que a su vez permite diseñar nuevas enzimas optimizadas.
MPI-DS, LMP
En primer lugar, la interfaz de la enzima y la molécula debe estar situada en su respectivo extremo más pequeño. De este modo, se puede conseguir un fuerte acoplamiento entre ambas. Por la misma razón, el cambio conformacional de la enzima no debe ser menor que el de la reacción. Por último, el cambio conformacional de la enzima tiene que producirse lo suficientemente rápido como para maximizar la fuerza motriz química de la reacción.
"Construimos nuestra investigación sobre dos pilares fundamentales", describe el planteamiento Ramin Golestanian, director del MPI-DS. "La conservación del momento y el acoplamiento entre las coordenadas de la reacción", prosigue. Así, los investigadores ampliaron la visión de una coordenada de reacción bidimensional clásica. Normalmente, los modelos de reacciones enzimáticas definen una barrera energética que hay que superar para que se produzca la reacción.
"Como en nuestro modelo también tenemos en cuenta la dinámica y el acoplamiento de las enzimas, vamos más allá de este concepto existente, considerando dos coordenadas de reacción", afirma Michalis Chatzittofi, primer autor del estudio. "En lugar de superar una barrera energética, ahora se pueden imaginar formas alternativas de sortearla tomando rutas alternativas", concluye.
Estos resultados proporcionan una nueva base para el diseño de máquinas moleculares, evitando el tedioso y técnicamente difícil planteamiento de simular la dinámica de cada átomo por separado.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original



Más noticias del departamento ciencias





















Noticias más leídas










Más noticias de nuestros otros portales




Contenido visto recientemente
MedImmune anuncia a los ganadores de la competición europea de abstract de investigación sobre el cáncer

Merck invierte 70 millones de euros en ampliar la fabricación de ADC para nuevas terapias contra el cáncer - Se triplica la capacidad de fabricación para satisfacer la creciente demanda mundial de conjugados anticuerpo-fármaco (ADC)
