Si possono dedurre tre regole d'oro per progettare enzimi ottimizzati per le reazioni chimiche
Istruzioni per l'assemblaggio degli enzimi
Annunci



In biologia, gli enzimi si sono evoluti nel corso di milioni di anni per guidare le reazioni chimiche. Gli scienziati del Max Planck Institute for Dynamics and Self-Organization (MPI-DS) hanno ora derivato regole universali per consentire la progettazione de novo di enzimi ottimali. Come esempio, hanno considerato la reazione enzimatica di rottura di un dimero in due molecole monomere. Considerando la geometria di un tale complesso enzima-substrato, hanno identificato tre regole d'oro che dovrebbero essere considerate per costruire un enzima funzionale.
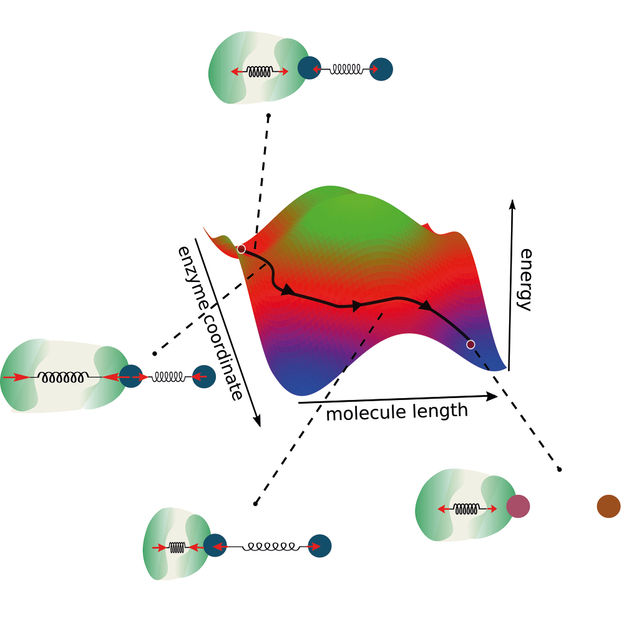
Un nuovo modello di reazioni enzimatiche estende la coordinata di reazione tradizionale considerando l'accoppiamento tra l'enzima e la molecola. In questo modo, si aprono strade alternative per le reazioni enzimatiche che, a loro volta, consentono la progettazione di nuovi enzimi ottimizzati.
MPI-DS, LMP
In primo luogo, l'interfaccia tra l'enzima e la molecola deve trovarsi all'estremità più piccola. In questo modo, è possibile ottenere un forte accoppiamento tra entrambi. Per lo stesso motivo, il cambiamento conformazionale dell'enzima non deve essere inferiore a quello della reazione. Infine, il cambiamento conformazionale dell'enzima deve avvenire abbastanza velocemente da massimizzare la forza chimica trainante della reazione.
"Abbiamo costruito la nostra ricerca su due pilastri principali", descrive Ramin Golestanian, direttore dell'MPI-DS. "La conservazione della quantità di moto e l'accoppiamento tra le coordinate della reazione", continua. In questo modo, i ricercatori hanno ampliato la visione di una classica coordinata di reazione bidimensionale. In genere, i modelli per le reazioni enzimatiche definiscono una barriera energetica che deve essere superata affinché la reazione abbia luogo.
"Poiché nel nostro modello consideriamo anche la dinamica e l'accoppiamento degli enzimi, andiamo oltre questo concetto esistente, considerando due coordinate di reazione", spiega Michalis Chatzittofi, primo autore dello studio. "Invece di superare una barriera energetica, ora si possono immaginare modi alternativi per aggirarla seguendo percorsi alternativi", conclude.
Questi risultati forniscono una nuova base per la progettazione di macchine molecolari, evitando l'approccio noioso e tecnicamente impegnativo di simulare la dinamica di ogni singolo atomo.
Nota: questo articolo è stato tradotto utilizzando un sistema informatico senza intervento umano. LUMITOS offre queste traduzioni automatiche per presentare una gamma più ampia di notizie attuali. Poiché questo articolo è stato tradotto con traduzione automatica, è possibile che contenga errori di vocabolario, sintassi o grammatica. L'articolo originale in Inglese può essere trovato qui.
Pubblicazione originale



Altre notizie dal dipartimento scienza

































