Three golden rules can be inferred to design optimized enzymes for chemical reactions
Assembly instructions for enzymes
Advertisement



In biology, enzymes have evolved over millions of years to drive chemical reactions. Scientists from the Max Planck Institute for Dynamics and Self-Organization (MPI-DS) now derived universal rules to enable the de novo design of optimal enzymes. As an example, they considered the enzymatic reaction of breaking a dimer into two monomer molecules. Considering the geometry of such an enzyme-substrate-complex, they identified three golden rules that should be considered to build a functional enzyme.
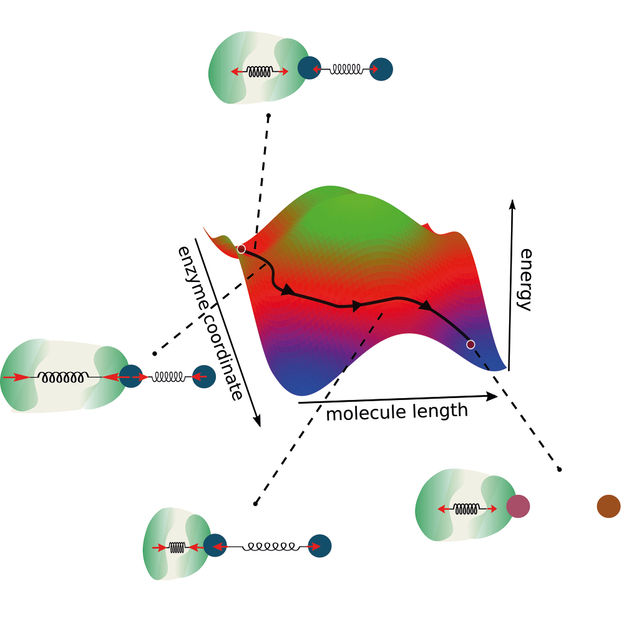
A new model of enzymatic reactions extends the traditional reaction coordinate by considering coupling between the enzyme and the molecule. This way, alternative ways for enzymatic reactions open which in turn enables the design of novel optimized enzymes.
MPI-DS, LMP
First, the interface of both enzyme and molecule should be located at their respective smaller end. This way, a strong coupling between both of them can be achieved. For the same reason, the conformational change in the enzyme should not be smaller than in the reaction. Finally, the conformational change of the enzyme has to take place fast enough to maximize the chemical driving force of the reaction.
“We built our research on two main pillars,” Ramin Golestanian, director of MPI-DS describes the approach. “Conservation of momentum and coupling between the reaction coordinates,” he continues. Thus, the researchers expanded the view of a classical 2-dimensional reaction coordinate. Typically, models for enzymatic reactions define an energy barrier that has to be overcome in order for the reaction to take place.
“As in our model we also consider the enzyme dynamics and coupling, we go beyond this existing concept, considering two reaction coordinates,” say Michalis Chatzittofi, first author of the study. “Instead of overcoming an energy barrier, one can now imagine alternative ways to bypass it by taking alternative routes,” he concludes.
These results provide a new basis for the design of molecular machines, avoiding the tedious and technically challenging approach to simulate the dynamics of each atom individually.
Original publication



Other news from the department science





















Most read news










More news from our other portals


























Last viewed contents
Thermo Fisher Scientific Opens New Manufacturing Facility in China - Expands Global Footprint, Local Production Capabilities to Address Increased Demand from Life Sciences Customers in China and Asia-Pacific Region
GSK and Theravance announce combination ICS/LABA Phase II results in the Relovair" development programme
Novexel’s NXL104/ceftazidime Combination Commences Phase II Clinical Trial

Albrecht Wiener joins Elementar as Head of Global Sales
