Podem deduzir-se três regras de ouro para conceber enzimas optimizadas para reacções químicas
Instruções de montagem para enzimas
Anúncios



Na biologia, as enzimas evoluíram ao longo de milhões de anos para conduzir reacções químicas. Cientistas do Instituto Max Planck de Dinâmica e Auto-Organização (MPI-DS) deduziram agora regras universais que permitem a conceção de novo de enzimas óptimas. Como exemplo, consideraram a reação enzimática de quebrar um dímero em duas moléculas de monómero. Considerando a geometria de um tal complexo enzima-substrato, identificaram três regras de ouro que devem ser consideradas para construir uma enzima funcional.
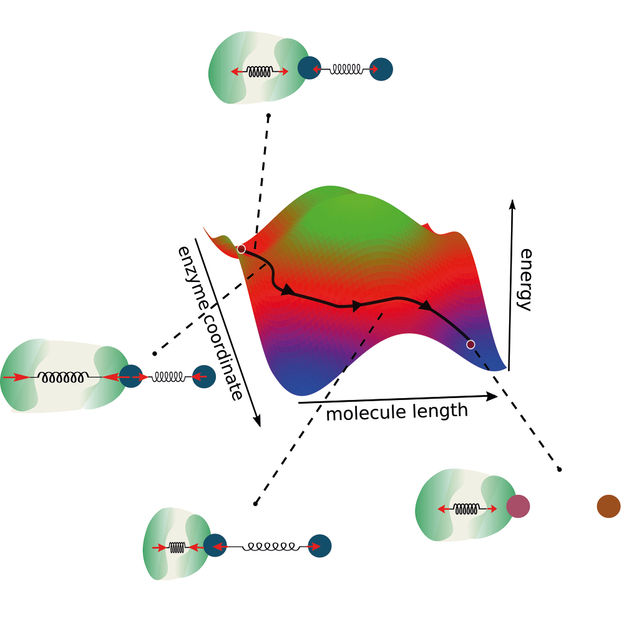
Um novo modelo de reacções enzimáticas alarga a coordenada de reação tradicional ao considerar o acoplamento entre a enzima e a molécula. Desta forma, abrem-se caminhos alternativos para as reacções enzimáticas que, por sua vez, permitem a conceção de novas enzimas optimizadas.
MPI-DS, LMP
Em primeiro lugar, a interface entre a enzima e a molécula deve estar localizada na sua extremidade mais pequena. Desta forma, é possível obter um forte acoplamento entre ambos. Pela mesma razão, a alteração conformacional da enzima não deve ser menor do que a da reação. Por último, a alteração conformacional da enzima tem de ser suficientemente rápida para maximizar a força motriz química da reação.
"Construímos a nossa investigação sobre dois pilares principais", descreve Ramin Golestanian, diretor do MPI-DS. "A conservação do momento e o acoplamento entre as coordenadas da reação", continua. Assim, os investigadores expandiram a visão de uma coordenada de reação bidimensional clássica. Normalmente, os modelos de reacções enzimáticas definem uma barreira energética que tem de ser ultrapassada para que a reação ocorra.
"Como no nosso modelo também consideramos a dinâmica e o acoplamento das enzimas, vamos além deste conceito existente, considerando duas coordenadas de reação", afirma Michalis Chatzittofi, primeiro autor do estudo. "Em vez de ultrapassar uma barreira energética, podemos agora imaginar formas alternativas de a contornar através de rotas alternativas", conclui.
Estes resultados fornecem uma nova base para a conceção de máquinas moleculares, evitando a abordagem tediosa e tecnicamente difícil de simular a dinâmica de cada átomo individualmente.
Observação: Este artigo foi traduzido usando um sistema de computador sem intervenção humana. A LUMITOS oferece essas traduções automáticas para apresentar uma gama mais ampla de notícias atuais. Como este artigo foi traduzido com tradução automática, é possível que contenha erros de vocabulário, sintaxe ou gramática. O artigo original em Inglês pode ser encontrado aqui.
Publicação original



Outras notícias do departamento ciência































