Una nueva herramienta facilita la interpretación clínica de la información genética
Anuncios



Equipos de investigación de Max Planck y Harvard desarrollan DeMAG, un nuevo método compartido como servidor web de código abierto (demag.org) para ayudar a interpretar mutaciones en genes de enfermedades y mejorar la toma de decisiones clínicas.
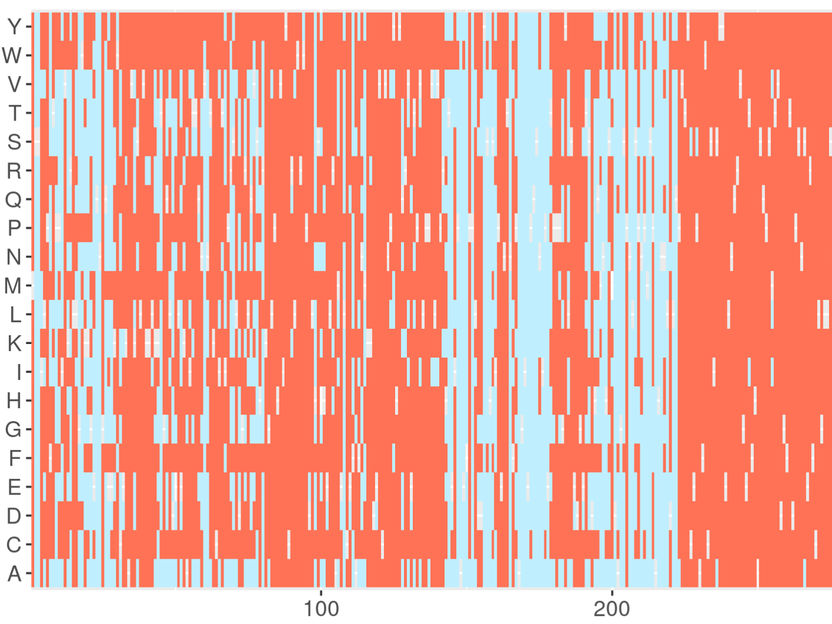
Captura de pantalla del servidor web de DeMAG. DeMAG predice las mutaciones benignas en azul claro y las patógenas en coral.
Agnes Toth-Petroczy, Nature Communications, 2023 / MPI-CBG
A pesar del creciente uso de la secuenciación genómica en la práctica clínica, la interpretación de mutaciones genéticas raras, incluso entre genes de enfermedades bien estudiadas, sigue siendo difícil. Los modelos predictivos actuales son útiles para interpretar esas mutaciones, pero son propensos a clasificar erróneamente las que no causan enfermedades, lo que contribuye a dar falsos positivos. Investigadores del Instituto Max Planck de Biología Celular Molecular y Genética (MPI-CBG) de Dresde, el Centro de Biología de Sistemas de Dresde (CSBD) en Alemania y la Facultad de Medicina de Harvard en Boston (EE.UU.) han desarrollado una herramienta llamada Deciphering Mutations in Actionable Genes (DeMAG) publicada en la revista Nature Communications. DeMAG es un servidor web de código abierto que ofrece una interpretación de los efectos de todas las posibles mutaciones de un solo aminoácido que podrían producirse en 316 genes clínicamente relevantes causantes de enfermedades para las que ya se dispone de diagnósticos y tratamientos preventivos. DeMAG proporciona a los profesionales médicos una herramienta que les permite evaluar con mayor precisión el efecto de las mutaciones en esos genes reduciendo la tasa de falsos positivos, lo que significa que las mutaciones menos benignas se predicen como patógenas. Como resultado, la herramienta puede apoyar la toma de decisiones clínicas.
En los últimos años, la secuenciación genómica se ha vuelto menos costosa y más avanzada. Por un lado, esto permite a los clínicos utilizar cada vez más la secuenciación con fines diagnósticos, al tiempo que permite a los científicos explorar más hipótesis de investigación. Por otro lado, muchas mutaciones detectadas no tienen una interpretación clínica clara. La incertidumbre sobre si una mutación causa una enfermedad puede ser estresante para los pacientes y generar carga psicológica, morbilidad y gastos sanitarios asociados al infradiagnóstico y al sobrediagnóstico. Aunque ya existen herramientas para predecir el impacto funcional de estas variantes, su rendimiento está sesgado debido a la escasez de datos clínicos, que dificulta la distinción entre variantes patogénicas (causantes de enfermedad) y benignas (neutras) dentro de un gen determinado y a menudo lleva a clasificar erróneamente como patogénicas mutaciones que no causan enfermedad. Resolver estas dificultades es fundamental para desarrollar un predictor fiable para aplicaciones clínicas.
El grupo de investigación de Agnes Toth-Petroczy en el MPI-CBG y el CSBD se asoció con Christopher Cassa, profesor adjunto de Medicina en la División de Genética del Brigham and Women's Hospital de la Facultad de Medicina de Harvard, e Ivan Adzhubei, investigador asociado del Departamento de Informática Biomédica de la Facultad de Medicina de Harvard, para desarrollar un modelo estadístico y el servidor web DeMAG que alcanza una gran precisión en la interpretación de mutaciones genéticas en genes patógenos. Para ello, los investigadores seleccionaron cuidadosamente mutaciones patógenas y benignas conocidas para entrenar el modelo. "Utilizamos bases de datos clínicas y de diversas poblaciones. Seleccionamos únicamente mutaciones cuya interpretación clínica está consensuada entre múltiples remitentes, como médicos y laboratorios de genética. Y también incluimos datos de ascendencias infrarrepresentadas en las bases de datos poblacionales actuales, como la coreana o la japonesa, para hacerlo aún más representativo y preciso", explica Federica Luppino, primera autora del trabajo de investigación y estudiante de doctorado en el grupo de Toth-Petroczy. DeMAG incluye una nueva función, la "puntuación de socios", que identifica grupos de aminoácidos en una proteína que comparten el mismo efecto clínico. Con la puntuación de socios, DeMAG aprovecha las relaciones entre aminoácidos basadas en la información evolutiva de los genomas de muchos organismos y la reciente revolución de la IA (Inteligencia Artificial) de predecir las formas 3D de las proteínas mediante el algoritmo AlphaFold desarrollado por Google DeepMind.
Agnes Toth-Petroczy, que supervisó el estudio, concluye: "Proporcionamos un marco básico para integrar datos clínicos y de proteínas con el fin de ayudar a evaluar el impacto de las mutaciones. Esperamos que nuestra herramienta y nuestro servidor web faciliten la evaluación del efecto de las variantes y la toma de decisiones clínicas. Además, las nuevas funciones desarrolladas pueden aplicarse a otros genes y organismos además de los humanos."
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original




Más noticias del departamento ciencias