Neue Methode zur Phylogenie und Klassifikation von Bakteriophagen und Archaeenviren
Wissenschaftler entwickeln Online-Werkzeug zur Bestimmung der Verwandtschaftsverhältnisse von Viren
Anzeigen



Viren, die Prokaryonten befallen, sind die mit Abstand häufigsten biologischen Strukturen auf diesem Planeten. In biogeochemischen Kreisläufen spielen sie eine entscheidende Rolle, in der Medizin sind sie Hoffnungsträger im Kampf gegen multiresistente Erreger. Auch die genetische Vielfalt dieser Viren ist gewaltig. Schätzungen zufolge stellen die bislang gut Tausend wissenschaftlich beschriebenen und offiziell anerkannten Arten nur einen winzigen Teil aller Arten von Bakterien- und Archaeen-Viren dar. Außerdem enthält ihre derzeitige Klassifikation einige Unstimmigkeiten, die unter anderem auf teils uneinheitlich angewandte, konventionelle Methoden zurückzuführen sind. Die Entwicklung und Anwendung von genomsequenzbasierten Methoden würde hier eine vielversprechende Verbesserung darstellen und ist in der aktuellen Literatur auch immer wieder gefordert worden. Dennoch gab es bisher kein Verfahren, um aus Genomsequenzen einen umfassenden, verlässlichen und gut interpretierbaren Stammbaum abzuleiten. Die Folgen können Unsicherheiten und Verwechslungen bei der Arbeit mit Viren sein.
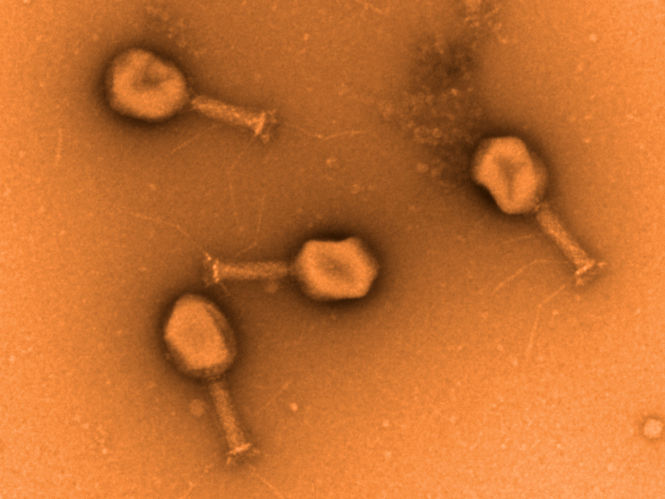
Transmissions-Elektronenmikroskopische Aufnahme von Phagen der Familie der Myoviridae gegen ESBL E. coli
Manfred Rohde, HZI
Jan Meier-Kolthoff und Markus Göker vom Leibniz-Institut DSMZ in Braunschweig schlagen nun einen neuen Ansatz zur Phylogenie und Klassifikation der Viren von Prokaryonten vor, der auf dem Gesamtgenom basiert, die Verwandtschaftsverhältnisse abbildet und Angaben zur Verlässlichkeit der Ergebnisse macht.
Grundlage des neuen Ansatzes ist eine bereits für die genombasierte Taxonomie von Bakterienarten etablierte Methode (GBDP – Genome BLAST Distance Phylogeny). Dabei werden komplette Genome paarweise verglichen und Distanzen berechnet. Meier-Kolthoff und Göker optimierten nun dieses Verfahren für die Anwendung bei Archaeenviren und Bakteriophagen, basierend auf einem umfangreichen Referenzdatensatz, der sich aus offiziell vom Internationalen Komitee für die Taxonomie von Viren (ICTV) anerkannten Virenarten zusammensetzte. Ihre Ergebnisse zeigen eine hohe Übereinstimmung mit der bestehenden Klassifikation. Die Methode stellt damit eine universelle, schnelle und zugleich zuverlässige Alternative zu bisherigen konventionellen Verfahren dar. Optimierte Grenzwerte für die Abgrenzung von Arten, Gattungen, Unterfamilien und Familien sind ebenfalls enthalten.
Mit der neuen Methode wurde darüber hinaus ein stark erweiterter Datensatz mit mehr als 4.000 Viren-Genomen aus öffentlich zugänglichen Datenbanken untersucht. Dies ermöglichte den beiden Bioinformatikern einen ersten Einblick in die noch unentdeckte Diversität, der eine große Zahl neuer Arten, Gattungen und (Unter-)Familien aufdeckte. Die Berechnung und der Vergleich der Viren-Genome setzte eine große parallele Rechenkapazität voraus. „Das war für uns nur möglich, weil wir die notwendigen Berechnungen mit Hilfe eines potenten DSMZ-Rechenclusters im Hochdurchsatzverfahren durchführen konnten“, erläutert Meier-Kolthoff.
Die neu entwickelte Methodik „VICTOR“ (Virus Classification and Tree Building Online Resource) steht auf der Webseite der DSMZ kostenlos für andere Wissenschaftler bereit. „Dort kann jeder mit wenigen Klicks Genomsequenzen von Viren hochladen – wahlweise Aminosäure- oder Nukleotiddaten – und erhält in der Regel nach wenigen Minuten Phylogenien mit statistischen Unterstützungswerten und einem Vorschlag zur Einteilung der Viren in Arten, Gattungen, Unterfamilien und Familien. Abgerundet wird das Angebot durch die Auslieferung von publikationsfertigem Text. Fast eine Art Rundum-Sorglos-Paket“, so Meier-Kolthoff. Die Initiatoren sind überzeugt, damit einiges zum Verständnis der Zusammensetzung der viralen Biosphäre beitragen zu können.
Die veröffentlichte Arbeit ist Teil des Sonderforschungsbereichs „Roseobacter“ (TRR 51), der einer Gruppe sehr häufiger Meeresbakterien gewidmet ist. Deren Phagen sind bislang weitgehend unerforscht. Mit Hilfe der neuen Methodik wollen die Projektpartner dies nun zielgerichtet angehen.
Originalveröffentlichung




Weitere News aus dem Ressort Wissenschaft






















































