Los rayos X miden la estructura de la proteína en el "corazón" del virus COVID-19.
La proteína de la proteasa tiene forma de corazón y funciona como una sola, permitiendo que el virus se replique y se extienda.
Anuncios



Un equipo de investigadores de los laboratorios nacionales Oak Ridge y Argonne del Departamento de Energía ha realizado las primeras mediciones de rayos X a temperatura ambiente de la principal proteasa del SARS-CoV-2, la enzima que permite que el virus se reproduzca.
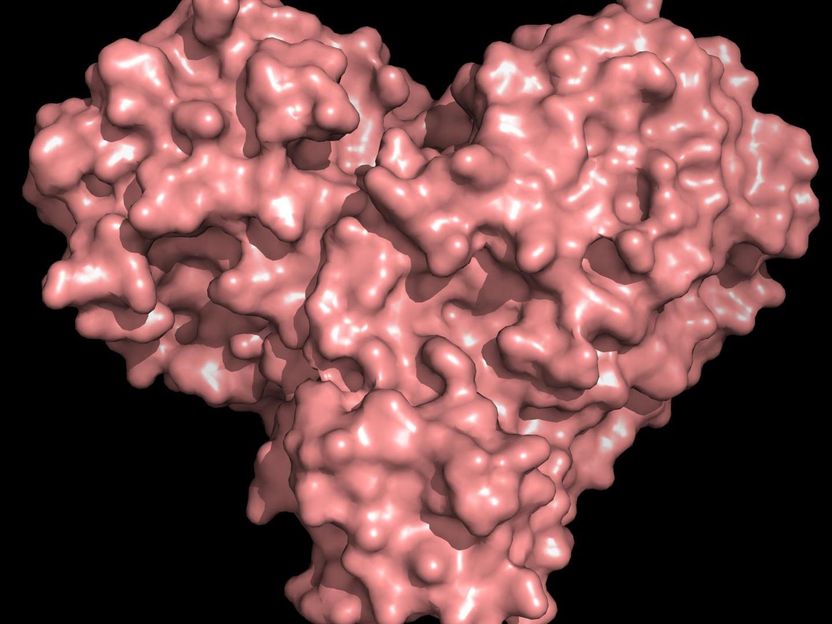
La proteína de la proteasa tiene forma de corazón y funciona como uno solo, permitiendo que el virus se replique y se extienda. Inhibir la proteasa bloquearía la reproducción del virus.
Andrey Kovalevsky/ORNL, U.S. Dept. of Energy
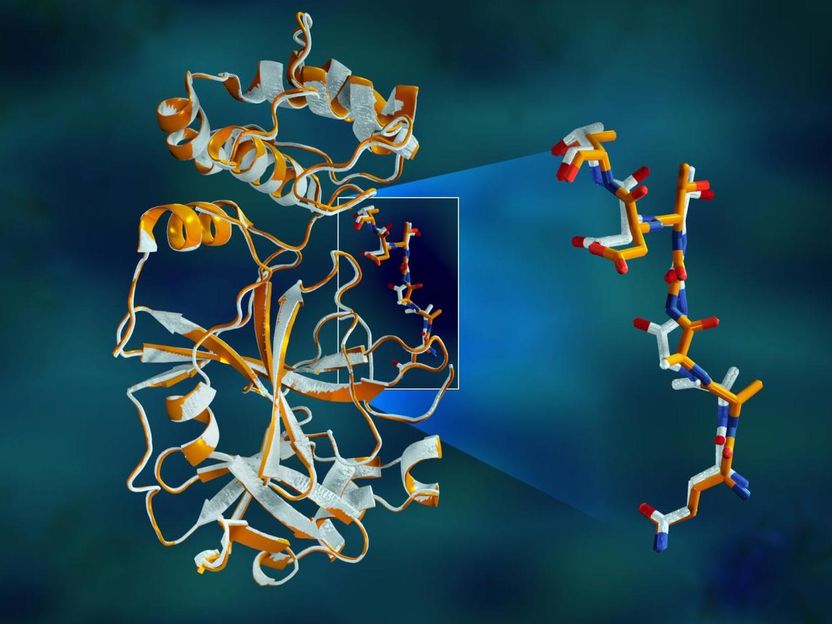
Los datos de rayos X superpuestos de la proteasa principal del SARS-CoV-2 muestran diferencias estructurales entre la proteína a temperatura ambiente (naranja) y la estructura congelada criogénicamente (blanca).
Jill Hemman/ORNL, U.S. Dept. of Energy
Las mediciones de rayos X marcan un importante primer paso en el objetivo final de los investigadores de construir un modelo 3D completo de la proteína enzimática. El modelo se utilizará para avanzar en las simulaciones de supercomputación destinadas a encontrar inhibidores de drogas para bloquear el mecanismo de replicación del virus y ayudar a poner fin a la pandemia COVID-19. Los resultados de sus investigaciones están a disposición del público y se han publicado en la revista Nature Communications.
El SARS-CoV-2 es el virus que causa la enfermedad COVID-19. El virus se reproduce expresando largas cadenas de proteínas que deben ser cortadas en menores longitudes por la enzima proteasa.
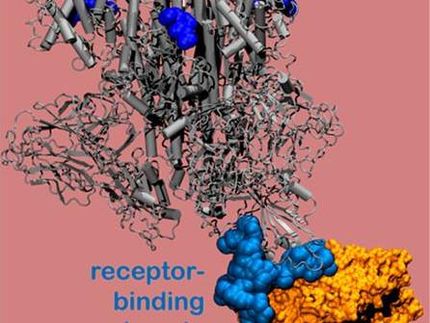
"La proteasa es indispensable para el ciclo de vida del virus. La proteína tiene la forma de un corazón de San Valentín, pero en realidad es el corazón del virus lo que le permite replicarse y propagarse. Si se inhibe la proteasa y se detiene el corazón, el virus no puede producir las proteínas que son esenciales para su replicación. Es por eso que la proteasa es considerada un blanco tan importante de la droga", dijo Andrey Kovalevsky de ORNL, autor correspondiente. Mientras que la estructura se conoce a partir de cristales preservados criogénicamente, "Esta es la primera vez que la estructura de esta enzima se ha medido a temperatura ambiente, lo que es significativo porque está cerca de la temperatura fisiológica en la que operan las células".
La construcción de un modelo completo de la estructura de la proteína requiere la identificación de cada elemento dentro de la estructura y cómo están dispuestos. Los rayos X son ideales para detectar elementos pesados como los átomos de carbono, nitrógeno y oxígeno. Debido a la intensidad de los rayos X en la mayoría de las instalaciones de sincrotrón a gran escala, las muestras biológicas normalmente deben congelarse criogénicamente a unos 100 K, o aproximadamente a menos 280 grados Fahrenheit, para soportar la radiación el tiempo suficiente para que se recojan los datos.
Para prolongar la vida útil de las muestras de proteínas cristalizadas y medirlas a temperatura ambiente, los investigadores del ORNL cultivaron cristales más grandes que los necesarios para los estudios con cristales de sincrotrón y utilizaron una máquina interna de rayos X que presenta un haz menos intenso.
"El cultivo de cristales de proteína y la recolección de datos es un proceso tedioso y que consume mucho tiempo. En el tiempo que típicamente toma preparar y enviar la muestra a un sincrotrón, fuimos capaces de hacer crecer los cristales, tomar las medidas y comenzar a analizar los datos", dijo Daniel Kneller de ORNL, el primer autor del estudio. "Y, cuando hay una pandemia con muchos científicos movilizándose para estudiar este problema, no hay un día libre".
La enzima proteasa consiste en cadenas de aminoácidos con un patrón repetitivo de átomos de nitrógeno-carbono que forman la espina dorsal de la proteína. Los grupos laterales de los bloques de construcción de aminoácidos, o "residuos", se extienden desde cada uno de los átomos de carbono de la columna vertebral central. La enzima se dobla en una forma 3D específica, creando bolsillos especiales donde una molécula de droga se adhiere.
El estudio reveló importantes disparidades estructurales entre las orientaciones de la columna vertebral y algunos de los residuos en las muestras a temperatura ambiente y criogénicas. La investigación sugiere que la congelación de los cristales puede introducir artefactos estructurales que podrían dar lugar a una comprensión menos precisa de la estructura de la proteasa.
Los resultados del equipo están siendo compartidos con los investigadores, dirigidos por el presidente del gobernador de la Universidad de Tennessee, ORNL, Jeremy Smith, que están llevando a cabo simulaciones de acoplamiento de drogas usando Summit en ORNL, la supercomputadora más rápida de la nación.
"Lo que los investigadores están haciendo en Summit es tomar compuestos de drogas conocidos y tratar de unirlos computacionalmente a la principal proteasa para el reprocesamiento de drogas, así como buscar nuevas pistas en otros potenciales candidatos a drogas", dijo el autor correspondiente de ORNL, Leighton Coates. "Nuestros datos de temperatura ambiente se están utilizando para construir un modelo más preciso para esas simulaciones y mejorar las actividades de diseño de drogas".
El siguiente paso de los investigadores para completar el modelo 3D de la proteasa principal del SARS-CoV-2 es utilizar la dispersión de neutrones en el Reactor de Isótopos de Alto Flujo de ORNL y en la Fuente de Neutrones de Espalación. Los neutrones son esenciales para localizar los átomos de hidrógeno, que desempeñan un papel fundamental en muchas de las funciones catalíticas y los esfuerzos de diseño de fármacos.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original
Anuncios



Más noticias del departamento ciencias
































