64 menschliche Genome als neue Referenz für die globale genetische Vielfalt
Referenzdaten bilden wichtige Grundlage: Ziel ist es, das individuelle Risiko für die Entstehung bestimmter Krankheiten wie Krebs abzuschätzen
Anzeigen



Eine internationale Forschungsgruppe, das „Human Genome Structural Variation Consortium (HGSVC), hat 64 menschliche Genome hochauflösend sequenziert. Zur Erfassung der genetischen Vielfalt der menschlichen Spezies wurden dafür Individuen aus der ganzen Welt einbezogen. Diese neuen Referenzdaten ermöglichen unter anderem bevölkerungsspezifische Studien zu genetischen Prädispositionen für menschliche Krankheiten sowie die Entdeckung komplexerer Formen genetischer Variationen, wie die 65 Autoren in der aktuellen Ausgabe der Fachzeitschrift Science berichten.
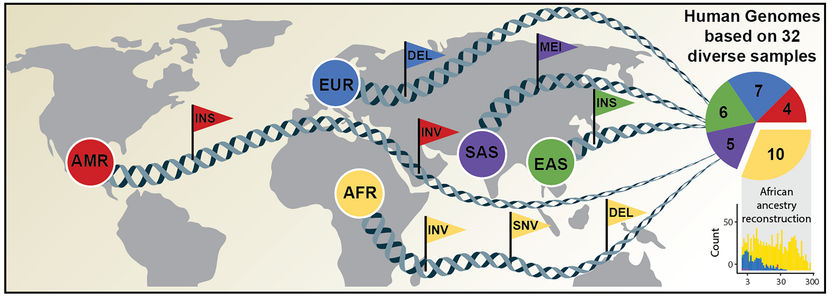
Umfassende Kartierung genetischer Variationen (symbolisiert durch Fähnchen) basierend auf der Analyse menschlicher Genome unterschiedlicher Abstammung.
University of Washington / David Porubsky
Im Jahr 2001 gab das „International Human Genome Sequencing Consortium“ den ersten Entwurf der Referenzsequenz des menschlichen Genoms bekannt. Am „Human Genome Project“ waren über elf Jahre lang mehr als 1.000 Forschende aus 40 Ländern beteiligt. Die damals vorgestellte Referenz erfasste aber nicht die gesamte Komplexität genetischer Variationen, denn sie repräsentierte kein einzelnes Individuum, sondern war ein Kompositum des Genoms von vielen Menschen.
Seit dem erfolgreichen Abschluss des Human Genome Project vor genau 20 Jahren haben Wissenschaftler zahlreiche Sequenzierungsprojekte durchgeführt, um genetische Unterschiede zwischen einem Individuum und dem Referenzgenom zu identifizieren und zu katalogisieren. Diese Unterschiede konzentrierten sich in der Regel auf kleine Veränderungen einzelner Basen und ließen größere genetische Veränderungen außer Acht. Aktuelle Technologien beginnen nun, größere Unterschiede – sogenannte strukturelle Varianten – wie Einfügungen von mehreren hundert Buchstaben zu erkennen und zu charakterisieren. Strukturelle Varianten beeinträchtigen mit höherer Wahrscheinlichkeit die Funktion von Genen, anders als die kleinen Veränderungen.
Ein internationales Forschungsteam hat jetzt einen Artikel in Science veröffentlicht, in dem ein neuer, wesentlich umfangreicherer Referenzdatensatz vorgestellt wird. Dieser wurde mithilfe einer Kombination aus fortschrittlichen Sequenzier- und Kartierungstechnologien gewonnen. Der neue Referenzdatensatz spiegelt 64 assemblierte menschliche Genome wider, die 25 verschiedene menschliche Populationen aus der ganzen Welt repräsentieren. Unter „(Genom-)Assemblierung“ versteht man dabei das Zusammensetzen eines Genoms aus einzelnen, von Sequenziergeräten gelesenen Fragmenten. Wichtig ist hierbei, dass jedes der Genome ohne Rückgriff auf das aktuelle Referenzgenom assembliert wurde. Somit wurden die genetischen Unterschiede verschiedener menschlicher Populationen besser erfasst.
Die Studie wurde von Wissenschaftlern des Europäischen Laboratoriums für Molekularbiologie in Heidelberg (EMBL), der Heinrich-Heine-Universität Düsseldorf (HHU), des Jackson Laboratory for Genomic Medicine in Farmington in Connecticut (JAX) und der University of Washington in Seattle (UW) geleitet.
„Mit diesen neuen Referenzdaten können genetische Unterschiede vor dem Hintergrund der globalen genetischen Variation mit bisher unerreichter Genauigkeit untersucht werden. Dies erleichtert die biomedizinische Bewertung der von einem Individuum getragenen genetischen Varianten“, betont der Co-Erstautor der Studie, Dr. Peter Ebert vom Institut für Medizinische Biometrie und Bioinformatik der HHU. Die Verteilung von genetischen Varianten kann sich durch spontane und kontinuierlich auftretende Veränderungen im Erbgut zwischen Bevölkerungsgruppen stark unterscheiden. Wird eine solche Mutation über viele Generationen weitergegeben, kann sie zu einer für diese Population spezifischen genetischen Variante werden.
Die neuen Referenzdaten bilden eine wichtige Grundlage, um das gesamte Spektrum genetischer Varianten in sogenannte genomweite Assoziationsstudien (engl. „genome wide association studies“) einzubeziehen. Ziel ist es, das individuelle Risiko für die Entstehung bestimmter Krankheiten wie Krebs abzuschätzen und die zugrundeliegenden molekularen Mechanismen zu verstehen. Dies wiederum kann als Grundlage für gezieltere Therapien und präventive Medizin genutzt werden.
Die nun vorliegenden Ergebnisse können weitere Anwendungen in der Präzisionsmedizin ermöglichen. Ein Beispiel: Die Wirksamkeit von Medikamenten kann von Individuum zu Individuum aufgrund des Genoms variieren. Die neuen Referenzdaten repräsentieren nun die gesamte Bandbreite verschiedener genetischer Varianten und umfassen menschliche Genome von großer Vielfalt. Mit Hilfe dieser neuen Ressource können möglicherweise auch neue Ansätze in der personalisierten Medizin entwickelt werden, bei denen die Auswahl von Therapien auf den individuellen genetischen Hintergrund eines Patienten zugeschnitten ist.
Die Studie baut auf einer neuen Methode auf, die von der Forschungsgruppe im vergangenen Jahr in Nature Biotechnology [DOI 10.1038/s41587-020-0719-5] veröffentlicht wurde. Mit dieser Technik gelingt es, die beiden Komponenten des Genoms einer Person genau zu rekonstruieren: eine, die vom Vater, eine, die von der Mutter geerbt wurde. Auf diese Weise eliminiert die Methode die potenziellen Verzerrungen, die sich beim Vergleich mit einem unvollkommenen Referenzgenom ergeben können.
Originalveröffentlichung

Anzeigen



Weitere News aus dem Ressort Wissenschaft

















































