O olhar mais profundo até à data sobre o genoma humano
Um novo recurso para a investigação do genoma a nível mundial

Em 2003, o Projeto do Genoma Humano (PGH) conseguiu mapear pela primeira vez mais de 90% do genoma humano. Posteriormente, o Projeto 1.000 Genomas (2007 - 2015), que reuniu dados de mais de 2.500 pessoas de diferentes populações e géneros, forneceu os primeiros conhecimentos sobre a diversidade do genoma humano. Agora - dez anos após a conclusão do projeto - um consórcio internacional de investigação, em colaboração com o Professor Dr. Tobias Marschall, bioinformático da Universidade Heinrich Heine de Düsseldorf (HHU), reanalisou as amostras utilizando técnicas de ponta. As descobertas resultantes são agora apresentadas em duas publicações no mesmo número da revista científica Nature. Mostram a diversidade do genoma humano com um nível de profundidade sem precedentes.
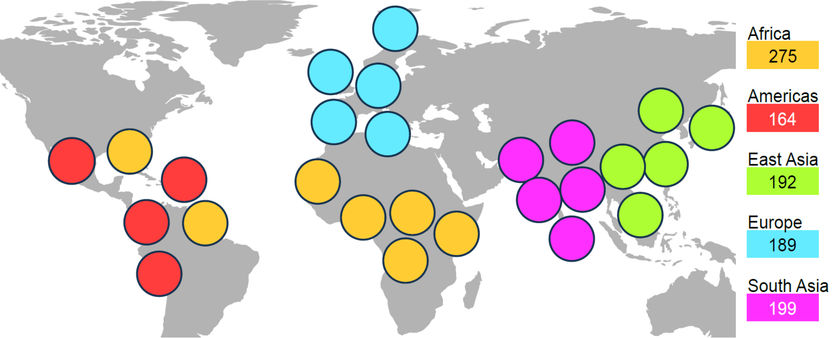
Os estudos analisaram conjuntos de dados do genoma de cinco continentes e 26 populações. Desta forma, foi possível mapear uma diversidade particularmente grande do genoma humano.
Copyright: Siegfried Schloissnig / HHU – Berit Meisenkothen
Com base no HGP, o objetivo do Projeto 1000 Genomas era sequenciar o genoma humano de uma secção transversal da população mundial. No final do projeto, em 2015, o objetivo tinha sido ultrapassado e tinham sido recolhidos dados de 2 500 pessoas de cinco continentes e 26 populações. Ambos os projectos deram contributos significativos para a compreensão do genoma humano.
Dez anos após a conclusão do projeto, um grupo de investigação internacional que envolveu a equipa dirigida pelo Professor Marschall (Instituto de Biometria Médica e Bioinformática) examinou agora o genoma humano em maior detalhe. Nos estudos agora publicados na revista científica Nature, os genomas recolhidos no âmbito do Projeto 1000 Genomas foram reanalisados utilizando tecnologias avançadas, que não estavam disponíveis em 2015.
O novo aspeto: Na altura do Projeto 1.000 Genomas, a sequenciação, ou seja, a determinação da sequência de letras do genoma humano, baseava-se em grande parte em segmentos curtos de ADN, que não eram suficientes para montar um genoma completo. Os novos métodos de sequenciação, ditos de leitura longa, permitem agora uma análise muito mais pormenorizada dos genomas. Estas tecnologias fornecem as sequências de segmentos de ADN mais longos de uma só vez, facilitando a identificação de diferenças genéticas entre indivíduos.
As chamadas variantes genéticas podem ocorrer de várias formas, como diferenças de um ou vários pares de bases - as letras - da sequência de ADN. No entanto, também podem ser mais extensas, por exemplo, quando segmentos de ADN mais longos são apagados, invertidos, repetidos ou adicionados em determinados indivíduos. Estes casos são também designados por variantes estruturais e desempenham um papel importante no desenvolvimento de uma série de doenças genéticas, incluindo síndromes genéticas raras e ainda inexplicadas.
O pangenoma: O mapeamento do genoma humano
Em 2023, o Projeto de Referência do Pangenoma Humano (HPRC), no qual o Professor Marschall também esteve envolvido, publicou um projeto de "pangenoma de referência", ou seja, um mapa da diversidade genética humana, baseado em 47 indivíduos. O objetivo é substituir no futuro o genoma de referência utilizado até à data por este projeto. Os dados do novo estudo também contribuirão para esse objetivo.
No primeiro dos estudos agora publicados (Schloissnig, Pani, et al.), foram sequenciados 1.019 genomas, o que torna esta coorte mais de 20 vezes maior do que os dados da HPRC. Este novo conjunto de dados de referência, significativamente maior, revela-se particularmente útil quando se estudam variantes estruturais que ocorrem com menor frequência na população. "Ter variantes numa coorte diversificada de indivíduos saudáveis é essencial para compreender melhor quais as variantes nos genomas dos doentes que podem ser a causa das doenças de que sofrem", afirma a Professora Dra. Dagmar Wieczorek (Instituto de Genética Humana da HHU), que também participou no estudo.
O segundo estudo (Logsdon, Ebert, Audano, Loftus, et al.) também alarga os conhecimentos existentes sobre o genoma humano. No entanto, o objetivo não era o número de genomas, mas sim a sequenciação dos genomas da forma mais completa possível. Foram examinadas 65 amostras, que também fazem parte do Projeto 1.000 Genomas, utilizando métodos de sequenciação altamente sofisticados. Os investigadores conseguiram reconstruir sequências completas do genoma (conhecidas como "T2T" ou "telómero a telómero") para 1161 cromossomas (39%). "Isto é particularmente digno de nota, uma vez que os cromossomas humanos podem conter centenas de milhões de pares de bases e só há alguns anos é que se conseguiu a primeira reconstrução completa de um genoma individual", afirma o bioinformático da HHU, Professor Dr. Alexander Dilthey (chefe da equipa de investigação do Instituto de Microbiologia Médica e Higiene Hospitalar), que também participou no estudo.
Além disso, os genomas completos permitiram agora compreender certas regiões, como os centrómeros, que não eram acessíveis através de métodos convencionais. Os centrómeros são os pontos em que as duas cromátides se ligam durante a divisão celular - formam a conhecida forma de X. A investigação sobre o significado e as consequências das variantes genéticas nos centrómeros tem sido limitada até à data. O novo estudo permite agora investigar a sua influência, por exemplo, em doenças imunitárias e cancros.
O Professor Dr. Jan Korbel do Laboratório Europeu de Biologia Molecular (EMBL) em Heidelberg, coautor de ambos os artigos, considera que a publicação simultânea dos dois estudos constitui um êxito especial: "Embora o primeiro estudo utilize métodos de sequenciação menos sofisticados, baseia-se numa coorte muito maior, enquanto o segundo estudo se baseia numa coorte mais pequena, mas utiliza métodos de sequenciação mais avançados. Isto permite-nos obter informações extremamente robustas e precisas sobre a variação dos nossos genomas".
Um novo recurso para a investigação do genoma a nível mundial
O Professor Marschall sublinha ainda que os resultados dos dois estudos não só fornecem informações importantes, como também aumentam significativamente a quantidade de dados disponíveis, o que terá um impacto positivo na investigação a longo prazo. "Estes estudos estabelecem um recurso extenso e clinicamente relevante, que pode agora ser utilizado por investigadores de todo o mundo para compreender melhor os mecanismos mutacionais que determinam a variação do genoma humano", afirma o Professor Marschall. "Este é um excelente exemplo de investigação em colaboração e de ciência aberta, que abre novas perspectivas na investigação do genoma e representa um passo em frente para um conhecimento mais completo do genoma humano. Estou confiante de que, com base nestas importantes descobertas, seremos capazes de identificar muitas ligações entre variantes genéticas estruturais e riscos de doença no futuro."
Os novos conjuntos de dados foram disponibilizados publicamente a investigadores de todo o mundo para análise e utilização.
Observação: Este artigo foi traduzido usando um sistema de computador sem intervenção humana. A LUMITOS oferece essas traduções automáticas para apresentar uma gama mais ampla de notícias atuais. Como este artigo foi traduzido com tradução automática, é possível que contenha erros de vocabulário, sintaxe ou gramática. O artigo original em Inglês pode ser encontrado aqui.
Publicação original
Siegfried Schloissnig, Samarendra Pani, Jana Ebler, Carsten Hain, Vasiliki Tsapalou, Arda Söylev, Patrick Hüther, Hufsah Ashraf, Timofey Prodanov, Mila Asparuhova, ...Tobias Rausch, Tobias Marschall, Jan O. Korbel; "Structural variation in 1,019 diverse humans based on long-read sequencing"; Nature, 2025-7-23
Glennis A. Logsdon, Peter Ebert, Peter A. Audano, Mark Loftus, David Porubsky, Jana Ebler, Feyza Yilmaz, Pille Hallast, Timofey Prodanov, DongAhn Yoo, Carolyn A. Paisie, William T. Harvey, Xuefang Zhao, Gianni V. Martino, Mir Henglin, ...Miriam K. Konkel, Jan O. Korbel, Charles Lee, Christine R. Beck, Evan E. Eichler, Tobias Marschall; "Complex genetic variation in nearly complete human genomes"; Nature, 2025-7-23
Outras notícias do departamento ciência