La mirada más profunda al genoma humano
Un nuevo recurso para la investigación del genoma en todo el mundo

Anuncios


En 2003, el Proyecto Genoma Humano (PGH) logró cartografiar por primera vez más del 90% del genoma humano. Posteriormente, el Proyecto de los 1.000 genomas (2007 - 2015), que recopiló datos de más de 2.500 personas de diferentes poblaciones y sexos, proporcionó una visión inicial de la diversidad del genoma humano. Ahora, diez años después de la conclusión del proyecto, un consorcio internacional de investigación, en colaboración con el Profesor Dr. Tobias Marschall, bioinformático de la Universidad Heinrich Heine de Düsseldorf (HHU), ha vuelto a analizar las muestras utilizando técnicas de vanguardia. Los resultados se presentan ahora en dos publicaciones en el mismo número de la revista científica Nature. Muestran la diversidad del genoma humano con un nivel de profundidad sin precedentes.
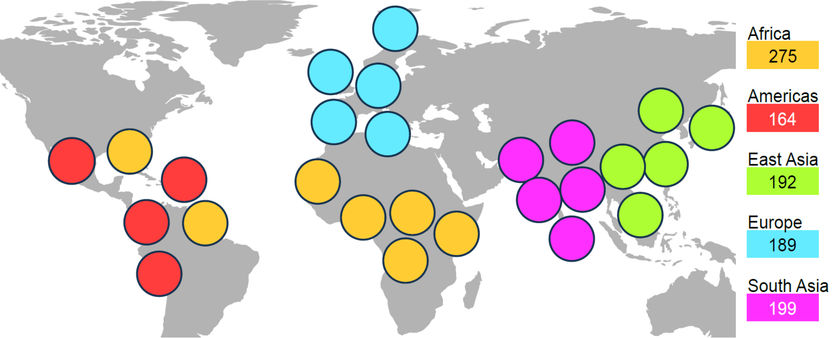
Los estudios analizaron conjuntos de datos genómicos de cinco continentes y 26 poblaciones. De este modo fue posible cartografiar una diversidad especialmente amplia del genoma humano.
Copyright: Siegfried Schloissnig / HHU – Berit Meisenkothen
Basándose en el HGP, el objetivo del Proyecto 1.000 Genomas era secuenciar el genoma humano de una muestra representativa de la población mundial. Al concluir el proyecto en 2015, se había superado el objetivo y se habían cotejado los datos de 2.500 personas de cinco continentes y 26 poblaciones. Ambos proyectos han contribuido significativamente a la comprensión del genoma humano.
Diez años después de la conclusión del proyecto, un grupo internacional de investigación en el que participa el equipo dirigido por el profesor Marschall (Instituto de Biometría Médica y Bioinformática) ha examinado ahora el genoma humano con más detalle. En los estudios publicados ahora en la revista científica Nature, los genomas recopilados en el marco del Proyecto de los 1.000 Genomas se volvieron a analizar utilizando tecnologías avanzadas, de las que no se disponía en 2015.

El nuevo aspecto: En la época del Proyecto 1.000 Genomas, la secuenciación, es decir, la determinación de la secuencia de letras en el genoma humano, se basaba en gran medida en segmentos cortos de ADN, que no bastaban para ensamblar un genoma completo. Los nuevos métodos de secuenciación denominados de lectura larga permiten ahora un análisis mucho más detallado de los genomas. Estas tecnologías proporcionan las secuencias de segmentos de ADN más largos en una sola pieza, lo que facilita la identificación de diferencias genéticas entre individuos.
Estas denominadas variantes genéticas pueden presentarse de diversas formas, como diferencias de uno o varios pares de bases -las letras- de la secuencia de ADN. Sin embargo, también pueden ser más extensas, por ejemplo, cuando se suprimen, invierten, repiten o añaden segmentos de ADN más largos en determinados individuos. Estos casos también se denominan variantes estructurales y desempeñan un papel importante en el desarrollo de diversas enfermedades genéticas, incluidos síndromes genéticos raros y aún inexplicados.
El pangenoma: La cartografía del genoma humano
En 2023, el Proyecto de Referencia del Pangenoma Humano (HPRC), en el que también participó el profesor Marschall, publicó un borrador del "pangenoma de referencia", es decir, un mapa de la diversidad genética humana, basado en 47 individuos. El objetivo es sustituir en el futuro el genoma de referencia utilizado hasta la fecha por este borrador. Los datos del nuevo estudio también contribuirán a ello.
En el primero de los estudios ahora publicados (Schloissnig, Pani, et al.), se secuenciaron 1.019 genomas, lo que hace que esta cohorte sea más de 20 veces mayor que los datos del HPRC. Este nuevo conjunto de datos de referencia, significativamente mayor, resulta especialmente útil a la hora de estudiar variantes estructurales que se dan con menos frecuencia en la población. "Disponer de variantes en una cohorte diversa de individuos sanos es esencial para comprender mejor qué variantes en los genomas de los pacientes pueden ser la causa de las afecciones que padecen", afirma la profesora Dra. Dagmar Wieczorek (Instituto de Genética Humana de la HHU), que también participó en el estudio.
El segundo estudio (Logsdon, Ebert, Audano, Loftus, et al.) también amplía los conocimientos existentes sobre el genoma humano. Sin embargo, en este caso no se centró en el número de genomas, sino en secuenciar los genomas de la forma más completa posible. Se examinaron 65 muestras, que también forman parte del Proyecto 1.000 Genomas, utilizando métodos de secuenciación muy sofisticados. Los investigadores pudieron reconstruir secuencias genómicas completas (conocidas como "T2T" o "telómero a telómero") de 1.161 cromosomas (39%). "Esto es especialmente destacable, ya que los cromosomas humanos pueden contener cientos de millones de pares de bases y hasta hace pocos años no se había logrado la primera reconstrucción completa de un genoma individual", afirma el bioinformático de la HHU Dr. Alexander Dilthey (jefe del equipo de investigación del Instituto de Microbiología Médica e Higiene Hospitalaria), que también participó en el estudio.
Además, los genomas completos han permitido ahora comprender ciertas regiones, como los centrómeros, a las que no se ha podido acceder con métodos convencionales. Los centrómeros son los puntos en los que se unen las dos cromátidas durante la división celular, formando la conocida X. La investigación sobre el significado y las consecuencias de las variantes genéticas en los centrómeros ha sido limitada hasta la fecha. El nuevo estudio permite ahora seguir investigando su influencia, por ejemplo, en trastornos inmunológicos y cánceres.
El Dr. Jan Korbel, catedrático del Laboratorio Europeo de Biología Molecular (EMBL) de Heidelberg y coautor de ambos trabajos, considera que la publicación simultánea de los dos estudios constituye un éxito especial: "Aunque el primer estudio utiliza métodos de secuenciación menos sofisticados, se basa en una cohorte mucho mayor, mientras que el segundo estudio se basa en una cohorte más pequeña pero utiliza métodos de secuenciación más avanzados. Esto nos permite obtener una visión extremadamente sólida y precisa de la variación de nuestros genomas".
Un nuevo recurso para la investigación del genoma en todo el mundo
El profesor Marschall también subraya que los resultados de los dos estudios no sólo aportan información importante, sino que también aumentan significativamente la cantidad de datos disponibles, lo que tendrá un impacto positivo en la investigación a largo plazo. "Estos estudios establecen un recurso amplio y médicamente relevante, que ahora puede ser utilizado por investigadores de todo el mundo para comprender mejor los mecanismos mutacionales que impulsan la variación del genoma humano", afirma el profesor Marschall. "Se trata de un ejemplo excepcional de investigación colaborativa y ciencia abierta, que abre nuevas perspectivas en la investigación del genoma y representa un paso hacia un conocimiento más completo del genoma humano. Confío en que, sobre la base de estos importantes hallazgos, podremos identificar en el futuro muchos vínculos entre variantes genéticas estructurales y riesgos de enfermedad."
Los nuevos conjuntos de datos se han puesto a disposición pública de investigadores de todo el mundo para su análisis y uso.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original
Siegfried Schloissnig, Samarendra Pani, Jana Ebler, Carsten Hain, Vasiliki Tsapalou, Arda Söylev, Patrick Hüther, Hufsah Ashraf, Timofey Prodanov, Mila Asparuhova, ...Tobias Rausch, Tobias Marschall, Jan O. Korbel; "Structural variation in 1,019 diverse humans based on long-read sequencing"; Nature, 2025-7-23
Glennis A. Logsdon, Peter Ebert, Peter A. Audano, Mark Loftus, David Porubsky, Jana Ebler, Feyza Yilmaz, Pille Hallast, Timofey Prodanov, DongAhn Yoo, Carolyn A. Paisie, William T. Harvey, Xuefang Zhao, Gianni V. Martino, Mir Henglin, ...Miriam K. Konkel, Jan O. Korbel, Charles Lee, Christine R. Beck, Evan E. Eichler, Tobias Marschall; "Complex genetic variation in nearly complete human genomes"; Nature, 2025-7-23




Más noticias del departamento ciencias