Erstes Open-Source-All-Atom-Modell des COVID-19 'S'-Proteins in voller Länge erstellt
Die Proteinstruktur erleichtert das Eindringen des Virus in die Wirtszellen und macht es zu einem Schlüsselziel für die Entwicklung von Impfstoffen und antiviralen Medikamenten
Anzeigen



Das Virus SARS-Coronavirus 2 (SARS-CoV-2) ist die bekannte Ursache der Coronavirus-Krankheit 2019 (COVID-19). Der "Spike" oder S-Protein erleichtert das Eindringen des Virus in die Wirtszellen.
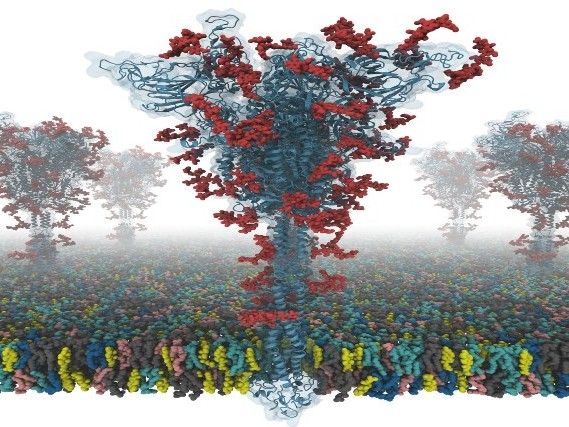
Ein Modell eines S-Proteins.
Dr. Yeolkyo Choi/Lehigh
Nun hat eine Gruppe von Forschern der Seoul National University in Südkorea, der University of Cambridge in Großbritannien und der Lehigh University in den USA zusammen gearbeitet, um die erste open-source, alle Atome umfassenden Modelle eines S-Proteins in voller Länge zu erstellen. Den Forschern zufolge ist dies von besonderer Bedeutung, da das S-Protein eine zentrale Rolle beim viralen Eintritt in die Zellen spielt und somit ein Hauptziel für die Entwicklung von Impfstoffen und antiviralen Medikamenten darstellt.
Die Einzelheiten finden Sie in der Veröffentlichung "Developing a Fully-glycosylated Full-length SARS-CoV-2 Spike Protein Model in a Viral Membrane", die online im The Journal of Physical Chemistry B veröffentlicht wurde.
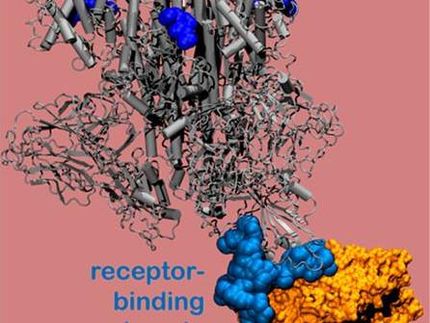
Ein Videodemo veranschaulicht, wie dieses Membransystem aus ihren SARS-CoV-2 S-Protein-Modellen aufgebaut werden kann. Das Modellbauprogramm ist frei zugänglich und kann auf der Homepage von CHARMM-GUI durch Klicken auf den Link COVID-19 Archiv gefunden werden.
CHARMM-GUI (GUI = Graphical User Interface) ist ein von Wonpil Im, Professor an der Fakultät für Biowissenschaften und Bioingenieurwesen der Lehigh University, entwickeltes Programm, das komplexe biomolekulare Systeme einfach, präzise und schnell simuliert. Im beschreibt es als ein "Computermikroskop", das es Wissenschaftlern ermöglicht, Wechselwirkungen auf molekularer Ebene zu verstehen, die auf andere Weise nicht beobachtet werden können.
"Unsere Modelle sind die ersten vollständig glykosylierten SARS-CoV-2-Spike (S)-Proteinmodelle in voller Länge, die anderen Wissenschaftlern zur Verfügung stehen", sagt Im. "Ich hatte das Glück, mit Dr. Chaok Seok von der Seoul National University in Korea und Dr. Tristan Croll von der University of Cambridge in Grossbritannien zusammenzuarbeiten. Unser Team verbrachte Tage und Nächte damit, diese Modelle sehr sorgfältig aus den bekannten Kryo-EM-Strukturanteilen aufzubauen. Die Modellierung war eine große Herausforderung, da es viele Regionen gab, in denen die einfache Modellierung keine qualitativ hochwertigen Modelle liefern konnte".
Wissenschaftler können die Modelle nutzen, um innovative und neuartige Simulationsforschung für die Prävention und Behandlung von COVID-19 zu betreiben, so Im.
Die Struktur des S-Proteins wurde mit Kryo-EM mit der RBD nach oben (PDB-ID: 6VSB) und mit der RBD nach unten (PDB-ID: 6VXX) bestimmt. Dieses Modell weist jedoch viele fehlende Rückstände auf. Daher modellierten sie zuerst die fehlenden Aminosäurereste und dann weitere fehlende Domänen. Darüber hinaus modellierten sie alle potenziellen Glykane (oder Kohlenhydrate), die an das S-Protein gebunden sind. Diese Glykane verhindern die Erkennung von Antikörpern, was die Entwicklung eines Impfstoffs erschwert. Sie bauten auch ein virales Membransystem aus einem S-Protein zur Simulation der Molekulardynamik auf.
Hinweis: Dieser Artikel wurde mit einem Computersystem ohne menschlichen Eingriff übersetzt. LUMITOS bietet diese automatischen Übersetzungen an, um eine größere Bandbreite an aktuellen Nachrichten zu präsentieren. Da dieser Artikel mit automatischer Übersetzung übersetzt wurde, ist es möglich, dass er Fehler im Vokabular, in der Syntax oder in der Grammatik enthält. Den ursprünglichen Artikel in Englisch finden Sie hier.
Originalveröffentlichung




Weitere News aus dem Ressort Wissenschaft





















































