Mathematik gegen Krebs
ONCOTYROL entwickelt statistische und rechenintensive Methoden zur Analyse von Gen-Netzwerken und fürs Drug Design
Anzeigen


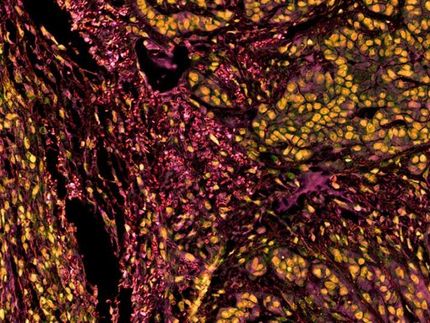
Komplexe Krankheiten wie Krebs haben in der Regel nicht nur eine molekulare Ursache. Vielmehr trägt eine Vielzahl von Faktoren zur Krankheitsentstehung bei. Um solche komplexen Zusammenhänge zu erkennen, ist es nötig, die Gesamtheit molekularer Vorgänge zu erfassen, beispielsweise welche Gene zu einem gewissen Zeitpunkt aktiv oder welche Stoffwechselabbauprodukte vorhanden sind. Moderne Hochdurchsatz-Analysemethoden sind dazu in der Lage: In automatisiert ablaufenden Massen-Experimenten liefern sie riesige Datenmengen. Bioinformatiker und Biostatistiker versuchen dann, in diesen Daten die Antwort auf biologische oder medizinische Fragen zu finden, beispielsweise: Welche Veränderung im Zusammenspiel der Gene unterscheidet einen Krebskranken von einem Gesunden? Häufig fehlen allerdings die mathematischen und statistischen Methoden, die für die Analyse komplexer biologischer Systeme geeignet sind. Im Rahmen von ONCOTYROL Center for Personalized Cancer Medicine konnten nun erste Erfolge bei der Entwicklung dieser dringend benötigten Verfahren erzielt werden.
ONCOTYROL widmet einen seiner fünf Forschungsbereiche diesen Fragestellungen. Unter Leitung von Prof. Armin Graber, Rektor der Privaten Landesuniversität UMIT in Hall, werden bioinformatische Methoden für die personalisierte Krebsmedizin erarbeitet. Einem seiner Kollegen, Prof. Matthias Dehmer, ist es nun gelungen, innovative Verfahren zur Analyse biologischer und biochemischer Netzwerke zu entwickeln.
Beispielweise stellen chemische Moleküle spezielle Netzwerke aus miteinander verbundenen Atomen dar. Dehmer und Co-Autoren haben eine neue mathematische Methode veröffentlicht (J. Chem Inf. Model. 2009, 49(7)), mit der sich Moleküle anhand ihrer Netzwerkstruktur aussagefähiger als bisher beschreiben und voneinander unterscheiden lassen. Die Besonderheit dieses Verfahrens ist, dass die Moleküle nicht notwendiger Weise als chemische Substanzen im Labor vorliegen müssen, vielmehr geschieht die eigentliche Analyse nur auf Basis Ihrer Netzwerkstruktur.
Solche Methoden sind für die Medikamentenentwicklung von großer Bedeutung, denn im Drug Design wird die Aktivität einer Substanz, beispielsweise seine Wirksamkeit oder Toxizität, anhand von Computermodellen vorhergesagt. Die Qualität dieser Vorhersagen ist aber von der möglichst eindeutigen Charakterisierung der Molekülstrukturen abhängig - und hier ist den ONCOTYROL-Forschern ein Quantensprung hin zu besonders eindeutigen Strukturmaßen gelungen. Eine ähnliche Fragestellung und die Untersuchung der strukturellen Verschiedenheit von biochemischen Netzen ist auch Thema einer neu eingereichten und bereits zur Veröffentlichung angenommenen Forschungsarbeit.
Weiter wurde gemeinsam mit Dehmers Kollegen, Dr. Frank Emmert-Streib von der Queen's University in Belfast, Leiter der Forschungsgruppe Computational Biology und Machine Learning am Center for Cancer Research and Cell Biology (CCRCB), ein Netzwerk von Zellzyklus-Genen mathematisch beschrieben und statistisch untersucht (BMC Systems Biology 2009, 3:76). Ein solches Gen-Netzwerk wird aus experimentellen Hochdurchsatz-Daten ermittelt. Es stellt dar, welche Gene „zusammenarbeiten“: Sie sind im Netzwerk miteinander verbunden. Der Zellzyklus ist der genetisch gesteuerte Rhythmus von Ruhephasen und Vermehrung von Zellen. Seine Fehlsteuerung trägt zur Krebsentstehung bei.
Beide Verfahren sind sinngemäß eng aneinander gekoppelt: Die Netzwerkanalyse hilft, das Zusammenspiel der Gene bei der Entstehungkomplexer Krankheiten wie Krebs besser zu verstehen - und die netzwerkbasierte Beschreibung von Molekülen unterstützt das Drug Design.



Weitere News aus dem Ressort Wissenschaft





















Meistgelesene News








Weitere News von unseren anderen Portalen





















Zuletzt betrachtete Inhalte
Bristol-Myers Squibb GesmbH - Wien, Österreich
Erstes Termitengenom entziffert
Deutsche Biotech Innovativ AG - Stadt Hennigsdorf, Deutschland
