Transplanted hematopoietic stem cells reverse damage caused by neuro-muscular disorder
Advertisement



Researchers at University of California San Diego School of Medicine report that a single infusion of wildtype hematopoietic stem and progenitor cells (HSPCs) into a mouse model of Friedreich's ataxia (FA) measurably halted cellular damage caused by the degenerative disease.
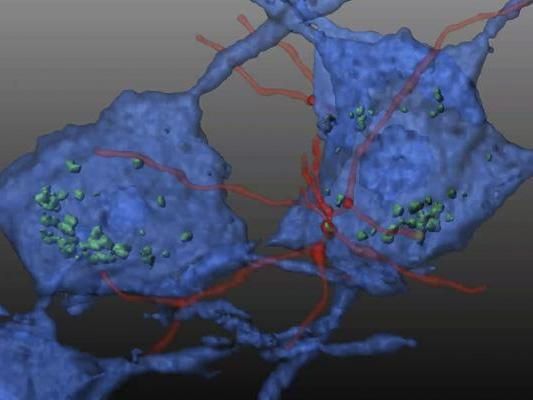
In this reconstituted schematic, hematopoietic stem cells (HSC) transplanted in a mouse model of Friedreich's ataxia differentiate into microglial cells (red) and transfer mitochondrial protein (green) to neurons (blue), preventing neurodegeneration.
Image courtesy of Stephanie Cherqui, UC San Diego School of Medicine
The findings suggest a potential therapeutic approach for a disease that currently is considered incurable.
Friedreich's ataxia is an inherited, degenerative neuromuscular disorder that initially impairs motor function, such as gait and coordination, but can lead to scoliosis, heart disease, vision loss and diabetes. Cognitive function is not affected. The disease is progressively debilitating, and ultimately requires full-time use of a wheelchair. One in 50,000 Americans has FA.

FA is caused by reduced expression of a mitochondrial protein called frataxin (FXN) due to a two mutated or abnormal copies of the FXN gene. In their study, Stephanie Cherqui, PhD, associate professor in the UC San Diego School of Medicine Department of Pediatrics, and colleagues used a transgenic mouse model that expresses two mutant human FXN transgenes, and exhibits the resulting progressive neurological degeneration and muscle weakness.
Human hematopoietic stem and progenitor cells (HSPCs), derived from bone marrow, have become a primary vehicle for efforts to replace or regenerate cells destroyed by a variety of diseases. Previous research by Cherqui and colleagues had shown that transplanting wildtype or normal mouse HSPCs resulted in long-term kidney, eye and thyroid preservation in a mouse model of cystinosis, another genetic disorder.
In this study, Cherqui's team transplanted wildtype HSPCs into an FA mouse model, reporting that the HSPCs engrafted and soon differentiated into macrophages in key regions of the mice's brain and spinal cord where they appeared to transfer wildtype FXN into deficient neurons and muscle cells.
"Transplantation of wildtype mouse HSPCs essentially rescued FA-impacted cells," said Cherqui, "Frataxin expression was restored. Mitochondrial function in the brains of the transgenic mice normalized, as did in the heart. There was also decreased skeletal muscle atrophy."
The scientists note that the mouse model is not perfect mirror of human FA. Disease progression is somewhat different and the precise pathology in mice is not fully known. However, Cherqui said the findings are encouraging and point toward a potential treatment for a disease that currently has none.
Original publication
Rocca, Celine J. and Goodman, Spencer M. and Dulin, Jennifer N. and Haquang, Joseph H. and Gertsman, Ilya and Blondelle, Jordan and Smith, Janell L. M. and Heyser, Charles J. and Cherqui, Stephanie; "Transplantation of wild-type mouse hematopoietic stem and progenitor cells ameliorates deficits in a mouse model of Friedreich’s ataxia"; Science Translational Medicine; 2017


Other news from the department science