Investigadores de la TU Graz abren nuevas vías para comprender las proteínas
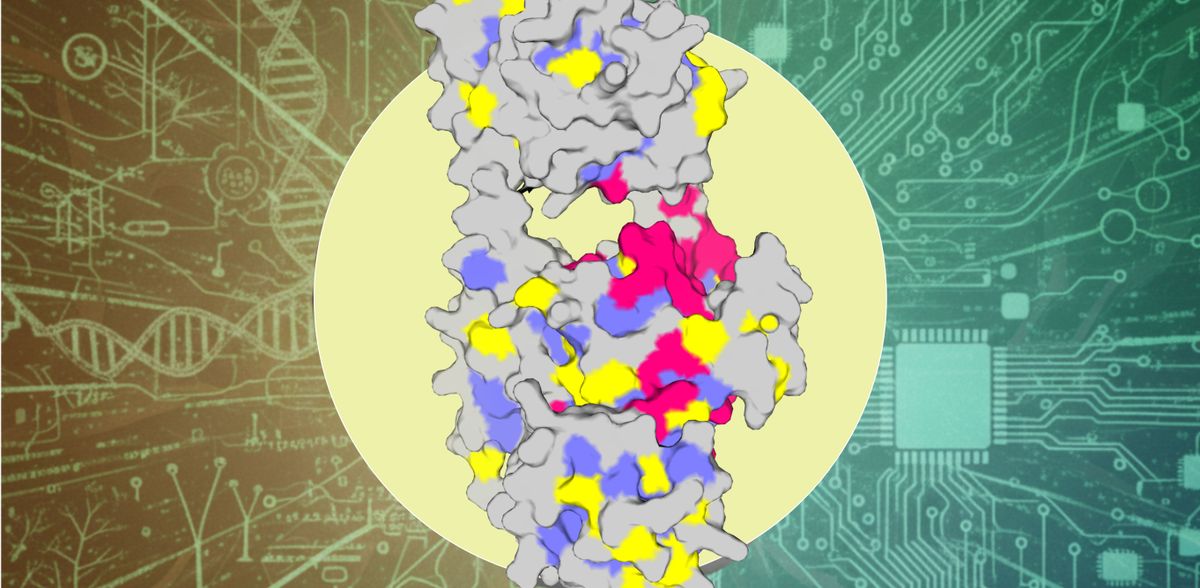
Gracias a un nuevo método que compara las secuencias de proteínas generadas por IA con las naturales, es posible determinar con mucha más precisión que antes los aminoácidos que regulan la función y la estructura.
Las proteínas son uno de los componentes básicos más importantes de la naturaleza y desempeñan un papel fundamental en los procesos biológicos de todos los organismos. Por ello, los científicos están muy interesados en comprenderlas con la mayor precisión posible. Como polímeros de diferentes aminoácidos, las proteínas pueden tener distintas estructuras tridimensionales y diversas funciones. Sin embargo, a menudo es difícil determinar qué aminoácidos influyen en la función proteica y cuáles en la estabilidad estructural.
Un equipo dirigido por Andreas Winkler y Oliver Eder, del Instituto de bioquímica de la Universidad Politécnica de Graz (TU Graz), ha logrado un gran avance con el método denominado "adaptabilidad función-estructura" (FSA), que acaba de publicar en la revista "Structure". La FSA compara secuencias de proteínas idealizadas generadas mediante aprendizaje automático con secuencias naturales que se han desarrollado a lo largo de millones de años de evolución. Esto permite identificar con una precisión sin precedentes los aminoácidos cruciales para la función y la estabilidad. Este conocimiento proporciona una base importante para la producción y modificación de proteínas y, por tanto, para el desarrollo de nuevos fármacos, para la mejora selectiva de proteínas en aplicaciones industriales y para una mejor comprensión de los cambios en las proteínas, por ejemplo en relación con la resistencia a los antibióticos.
Comprender mejor los componentes básicos de la vida
"Como bioquímicos, queremos entender cómo han evolucionado las proteínas en la naturaleza y averiguar qué aminoácidos son relevantes para funciones específicas", explica Andreas Winkler. "Para ello, combinamos lo que la naturaleza ha conservado durante la evolución con lo que un modelo de IA considera relevante para la estabilidad y estructura de una proteína. Esta combinación de millones de años de historia evolutiva y la tecnología más avanzada simplifica enormemente el análisis y la comprensión de las proteínas."
Para su método, el equipo utilizó el modelo de aprendizaje profundo ProteinMPNN, que genera nuevas secuencias de proteínas con el objetivo de garantizar que adopten una estructura tridimensional estable predeterminada. Los investigadores compararon estas secuencias con las de las proteínas naturales. Como sistema de prueba, se utilizó la familia de proteínas bacteriofitocromo, que en la naturaleza sirve como fotorreceptor para algunas bacterias y desempeña un papel central en la percepción de influencias ambientales como la luz. El nuevo método de análisis reveló que si un aminoácido está repetidamente representado en las secuencias naturales, pero no parece ser significativo para ProteinMPNN, esto indica un papel funcional. Sin embargo, si está muy presente en ambas colecciones de secuencias, es un indicio de importancia estructural.
Validación en el laboratorio
Para su planteamiento, los investigadores tuvieron que agrupar los aminoácidos en función de sus propiedades químicas con el fin de comparar estadísticamente las proteínas naturales y las generadas por IA. Esto permitió clasificar los aminoácidos en tres categorías: "funcionales" (importantes para el papel específico de la proteína), "estructurales" (relevantes para la estabilidad y el plegamiento) y "adaptables" (una tercera categoría que aún requiere más investigación). El equipo validó los resultados mediante amplios experimentos de laboratorio en los que pudieron influir en las propiedades funcionales de las proteínas introduciendo cambios específicos en los aminoácidos clasificados correspondientemente. Esto permitió, por ejemplo, influir significativamente en la percepción de la luz del sistema de prueba de los fotorreceptores. La comparación con residuos funcionales ya conocidos en la bibliografía también confirmó el elevado porcentaje de aciertos del nuevo método de análisis.
"En el pasado, a menudo se necesitaban varios meses o incluso años de trabajo preparatorio y de laboratorio para llevar a cabo un análisis como éste", afirma Oliver Eder. "Ahora, el trabajo preliminar para identificar secuencias de proteínas naturales potencialmente interesantes es posible para una nueva proteína en el plazo de una semana. Y como nuestro método nos permite filtrar previamente los aminoácidos funcionales de forma mucho más específica, no tenemos que dedicar tanto tiempo en el laboratorio a las pruebas y la caracterización". Como el método puede aplicarse en principio a todas las clases de proteínas, ahora podemos apreciar los intrincados detalles del funcionamiento de las proteínas de una forma más específica".
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original

Más noticias del departamento ciencias