I ricercatori del TU Graz aprono nuove strade alla comprensione delle proteine
Annunci


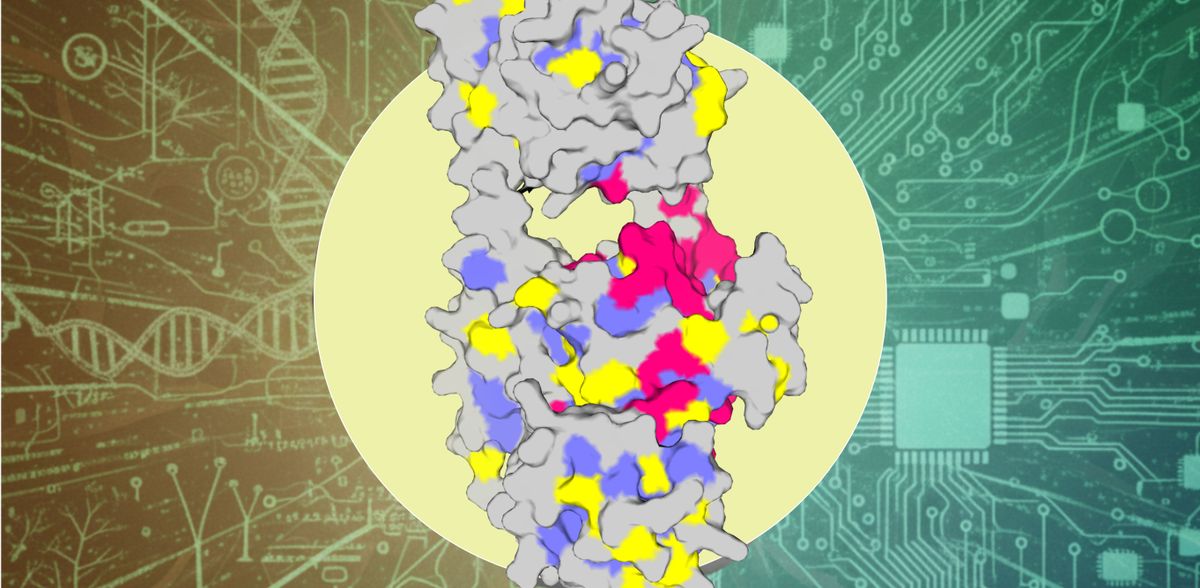
Grazie a un metodo di recente sviluppo che confronta le sequenze proteiche generate dall'intelligenza artificiale con quelle presenti in natura, è possibile determinare con maggiore precisione gli amminoacidi che regolano la funzione e la struttura.
Le proteine sono tra i più importanti elementi costitutivi della natura e svolgono un ruolo centrale nei processi biologici di tutti gli organismi. Di conseguenza, gli scienziati sono interessati a comprenderle nel modo più preciso possibile. Essendo polimeri di diversi amminoacidi, le proteine possono avere diverse strutture tridimensionali e varie funzioni. Tuttavia, è spesso difficile determinare quali aminoacidi influenzino la funzione delle proteine e quali la stabilità strutturale.
Utilizzando il cosiddetto approccio Function-Structure-Adaptability (FSA), un team guidato da Andreas Winkler e Oliver Eder dell'Istituto di Biochimica dell'Università di Tecnologia di Graz (TU Graz) ha raggiunto una svolta, ora pubblicata sulla rivista "Structure". L'FSA confronta sequenze proteiche idealizzate generate con l'apprendimento automatico con sequenze naturali che si sono sviluppate in milioni di anni di evoluzione. Ciò consente di identificare con una precisione senza precedenti gli amminoacidi cruciali per la funzione e la stabilità. Questa conoscenza fornisce una base importante per la produzione e la modifica delle proteine e quindi per lo sviluppo di nuovi farmaci, per il miglioramento mirato delle proteine nelle applicazioni industriali e per una migliore comprensione dei cambiamenti delle proteine, ad esempio in relazione alla resistenza agli antibiotici.
Capire meglio i mattoni della vita
"Come biochimici, vogliamo capire come si sono evolute le proteine in natura e quindi scoprire quali amminoacidi sono importanti per funzioni specifiche", spiega Andreas Winkler. Per farlo, abbiamo combinato ciò che la natura ha conservato durante l'evoluzione con ciò che un modello di intelligenza artificiale considera rilevante per la stabilità e la struttura di una proteina". Questa combinazione di milioni di anni di storia evolutiva e della tecnologia più recente semplifica notevolmente l'analisi e la comprensione delle proteine".
Per il suo metodo, il team ha utilizzato il modello di deep learning ProteinMPNN, che genera nuove sequenze proteiche con l'obiettivo di garantire che adottino una struttura tridimensionale stabile predeterminata. I ricercatori hanno confrontato queste sequenze con quelle delle proteine naturali. Come sistema di prova è stata utilizzata la famiglia di proteine del batteriofotocromo, che in natura funge da fotorecettore per alcuni batteri e svolge un ruolo centrale nella percezione delle influenze ambientali come la luce. Il nuovo metodo di analisi ha rivelato che se un amminoacido è ripetutamente rappresentato nelle sequenze naturali, ma non sembra essere significativo per la ProteinMPNN, ciò indica un ruolo funzionale. Tuttavia, se è fortemente presente in entrambe le collezioni di sequenze, ciò indica un significato strutturale.
Convalida in laboratorio
Per il loro approccio, i ricercatori hanno dovuto raggruppare gli aminoacidi in base alle proprietà chimiche, per poi confrontare statisticamente le proteine naturali e quelle generate dall'intelligenza artificiale. In questo modo è stato possibile classificare gli amminoacidi in tre categorie: "funzionali" (importanti per il ruolo specifico della proteina), "strutturali" (rilevanti per la stabilità e il ripiegamento) e "adattabili" (una terza categoria che richiede ancora ulteriori ricerche). L'équipe ha convalidato i risultati mediante esperimenti di laboratorio approfonditi, durante i quali è stata in grado di influenzare le proprietà funzionali delle proteine apportando modifiche specifiche agli aminoacidi classificati in modo corrispondente. In questo modo è stato possibile, ad esempio, influenzare in modo significativo la percezione della luce del sistema di test dei fotorecettori. Anche il confronto con i residui funzionali già noti in letteratura ha confermato l'alto tasso di successo del nuovo metodo di analisi.
"In passato, spesso erano necessari diversi mesi o addirittura anni di lavoro preparatorio e di laboratorio per effettuare un'analisi di questo tipo", spiega Oliver Eder. "Il lavoro preliminare per identificare sequenze di proteine naturali potenzialmente interessanti è ora possibile per una nuova proteina nel giro di una settimana. E poiché il nostro metodo ci permette di pre-filtrare gli amminoacidi funzionali in modo molto più specifico, non dobbiamo dedicare molto tempo in laboratorio ai test e alla caratterizzazione". Poiché il metodo può essere applicato in linea di principio a tutte le classi di proteine, ora possiamo apprezzare i dettagli intricati del funzionamento delle proteine in modo più mirato".
Nota: questo articolo è stato tradotto utilizzando un sistema informatico senza intervento umano. LUMITOS offre queste traduzioni automatiche per presentare una gamma più ampia di notizie attuali. Poiché questo articolo è stato tradotto con traduzione automatica, è possibile che contenga errori di vocabolario, sintassi o grammatica. L'articolo originale in Inglese può essere trovato qui.
Pubblicazione originale




Altre notizie dal dipartimento scienza

































