Estudio de cientos de proteínas en paralelo
Primer método altamente escalable desarrollado para vigilar los niveles de proteínas y las localizaciones en las células
Anuncios



Los investigadores del CeMM han desarrollado un método altamente escalable que permite el estudio de cientos de proteínas en paralelo con el fin de monitorizar los cambios de sus niveles y su localización en la célula. Esta novedosa estrategia es una notable contribución, no sólo al desarrollo de fármacos para futuros tratamientos contra enfermedades como el cáncer, sino también a nuestra comprensión y conocimiento general de la dinámica de los proteomas.
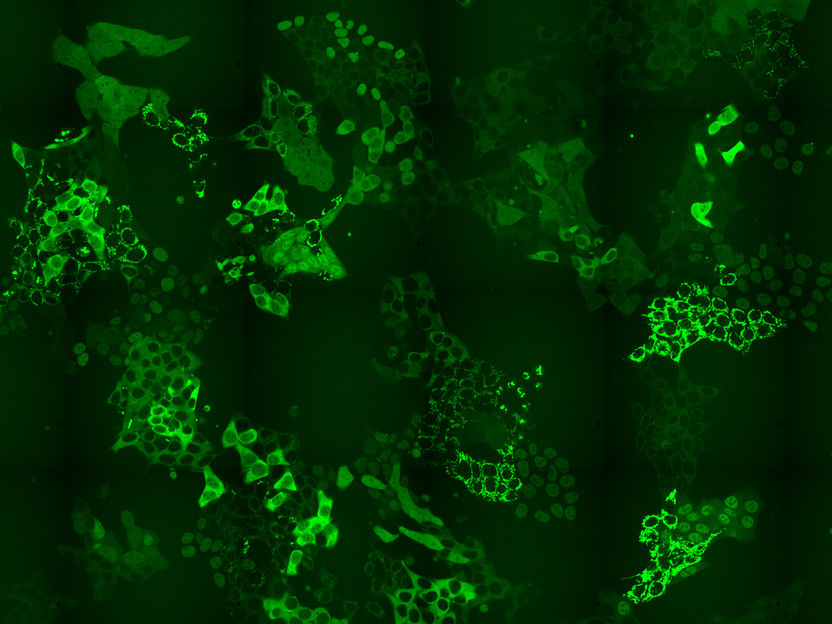
El conjunto de células que expresan cientos de diferentes proteínas de fusión GFP
Andreas Reicher
Las proteínas son grandes moléculas en la célula, y son necesarias para la estructura, función y regulación de los tejidos y órganos del cuerpo. Son responsables de casi todas las tareas de la vida celular y pueden ser tan diversas como las funciones a las que sirven. Los niveles de proteína y su localización dentro de la célula regulan aspectos importantes de muchos procesos celulares y pueden convertirse en objetivos importantes para el tratamiento con drogas. Por ejemplo, la abundancia de proteínas puede aumentar o disminuir mediante la intervención terapéutica, por medio de drogas que afectan a la producción y degradación de proteínas en la célula. Las proteínas también pueden desplazarse entre diferentes compartimentos celulares y, de ese modo, modificar sus funciones. Otras proteínas pueden unirse a lugares distintos en respuesta a estímulos externos, como las zonas en que se producen daños en el ADN.
Tradicionalmente, los científicos utilizan una etiqueta fluorescente para etiquetar las proteínas individuales y estudiar sus funciones en la célula. Una proteína verde fluorescente (GFP) se fusiona a uno de los extremos de una determinada proteína que querían estudiar. Esta fusión de la proteína se expresa entonces en la célula, y a través del microscopio de fluorescencia pueden observar las células que expresan la proteína etiquetada. Este método permite estudiar muchas perturbaciones, como diferentes dosis de medicamentos, de manera resuelta en el tiempo para una sola proteína. Por el contrario, la espectrometría de masas no era adecuada para estudiar y monitorizar estas perturbaciones celulares en el proteoma, el complemento entero de las proteínas, a un alto nivel escalable en un punto específico en el tiempo de forma imparcial.
Andreas Reicher y Anna Koren del grupo del investigador principal del CeMM Stefan Kubicek han desarrollado una estrategia novedosa que permite, por primera vez, observar y caracterizar esos cambios en un número muy elevado de proteínas en paralelo. Este método puede utilizarse no sólo para describir y comprender mejor los efectos de ciertos fármacos conocidos en las células, sino también para descubrir nuevos tratamientos farmacológicos que funcionan afectando y modulando los niveles o las localizaciones de las proteínas en las células.
Los investigadores del CeMM han diseñado un método para superar el cuello de botella en el etiquetado intrínseco basado en el CRISPR-CAS9: que es necesario desarrollar métodos que hagan brillar una luz sobre todo el proteoma, o una parte sustancial del mismo y no sólo una proteína a la vez. Para superar este problema, los investigadores del CeMM diseñaron un método para generar pools celulares que contienen cientos de proteínas etiquetadas, y en cada célula se etiquetó una proteína diferente con GFP. Estos pools celulares fueron expuestos a un degradador químico PROTAC de BRD4, un regulador transcripcional que juega un papel clave durante la embriogénesis y el desarrollo del cáncer. Luego, los investigadores, utilizando microscopía de lapso de tiempo, observaron si había algún cambio en los niveles o en la localización subcelular de cualquiera de las proteínas etiquetadas en el pool celular en respuesta al tratamiento aplicado. Es importante señalar que la estrategia de marcado CRISPR-Cas9 que aplicaron les permitió identificar qué proteínas cambiaban de localización mediante el uso de la secuenciación in situ de todo el conjunto de células. De esta manera, confirmaron los objetivos conocidos de esta droga pero también revelaron cambios inesperados. En particular para las perturbaciones de la señalización del BRD4, pudieron informar de cambios en las localizaciones de seis proteínas que anteriormente no habían sido reconocidas por ningún otro método de alto rendimiento. Por último, también mostraron que el método revela cambios esperados y novedosos en la localización de las proteínas como respuesta al tratamiento con la droga aprobada para el cáncer, el metotrexato.
El investigador principal del CeMM, Stefan Kubicek, explica: "Nuestro estudio describe una tecnología que no sólo aplica por primera vez el marcaje por intrusión a un pool de genes, sino que también se optimiza significativamente en los tres pasos - marcaje por intrusión, imágenes celulares y secuenciación in situ - para permitir el proceso de la manera más eficaz. Este método aplicado a las bibliotecas químicas y a las moléculas candidatas es particularmente poderoso para desarrollar y caracterizar profundamente las drogas, incluyendo la inducción e inhibición de las interacciones proteína-proteína y la degradación química. La estrategia descrita acelerará potencialmente el descubrimiento de drogas y tendrá un gran impacto en el estudio de la dinámica de los proteomas globales y subcelulares".
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original

Anuncios



Más noticias del departamento ciencias
































