Bungled cell division drives cancer cells of childhood tumors to death
Cancer researchers have discovered a mechanism that can activate the self-destruction of cancer cells of childhood tumors
Advertisement



A drug combination already being tested in other cancers caused the cancer cells to accumulate errors in their genetic material in mice, causing the bone and soft tissue tumors to shrink in the laboratory. The scientists from the Hopp Children's Cancer Center Heidelberg (KiTZ), Heidelberg University Hospital (UKHD) and the German Cancer Research Center (DKFZ) also showed that a key molecule of cell division could be used as a new biomarker to select patients who would benefit from this therapy.
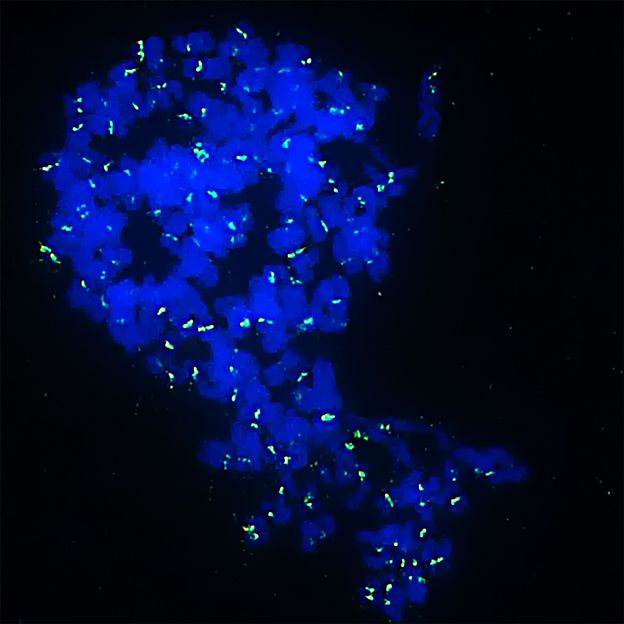
Inhibitors cause Ewing sarcoma cells to become monster cells with multiple sets of chromosomes (in blue) and then die. In green you can see the site of the chromosomes that holds both halves of the chromosomes together.
Jing Li/KiTZ
Normally, genetically degenerated cells are driven to suicide by a cellular program, preventing the development of cancer by their "voluntary" death. However, cancer cells manage to escape the so-called programmed cell death, also known as apoptosis, and continue to divide. Their unstable genetic material even gives them growth advantages and makes them resistant to therapy.
In contrast, in certain childhood cancers, such as Ewing sarcoma, the genome behaves remarkably quietly and few such genomic and chromosomal rearrangements are found. Ewing sarcomas are highly aggressive tumors that can form in bone or soft tissues and occur mainly in children and adolescents. "Many standard therapies fail, especially in this type of cancer, so new treatment options are urgently needed," says Thomas Grünewald, head of the Translational Pediatric Sarcoma Research Group at KiTZ and the department of the same name at DKFZ.
Together with scientists from Ludwig-Maximilians-Universität (LMU) Munich and the German Consortium for Translational Cancer Research (DKTK), Grünewald's team looked for a way to take advantage of the genetic stability of childhood cancer cells to drive them to death. "Children's cancer cells still respond to programmed cell death signals. Our hope was therefore that we only had to cause enough chaos in the cells' genome for them to initiate their self-destruction," Grünewald explains.
The scientists had already targeted a suitable molecular weak point in earlier work: The protein PRC1. "PRC1 contributes to the stable division of tumor cells and is increasingly produced by a mutated cancer gene in Ewing sarcoma," explains the first author of the study, Jing Li, who intensively studied the molecular interaction of the mutated gene with PRC1.
By combining two inhibitors that are already being tested in clinical trials against other cancers, the scientists succeeded in specifically disrupting PRC1 activity and thus the cell division of the cancer cells. The newly formed cancer cells accumulated more hereditary defects and formed multiple sets of chromosomes, which led to a bizarre appearance and gave rise to cells with huge nuclei. Tumors with these "monster cells" shrank even if the cancer cells had previously become resistant to therapy, the study in mice showed. The abnormal cell division and uneven chromosome distribution caused the cells to get their apoptosis machinery working again. However, this was only the case in tumors with high levels of PRC1.
"Previous results actually initially suggested that the drug combination used here was rather unpromising in patients with Ewing sarcoma," says Grünewald. "The new data now give us hope that it may well be effective in patients with high PRC1 activity. It shows once again how important it is to adapt cancer therapies to the molecular background of the tumor. "The scientists now want to test whether the PRC1 level is suitable as a reliable biomarker with further investigations and, if successful, develop it further in a clinical trial.
Original publication



Other news from the department science























































