Cancer research: The genetic context is crucial
Advertisement



An LMU team has uncovered a mechanism behind the variability in the clinical course of Ewing sarcoma. The interaction between the acquired driver mutation and the germline genetic context in which it occurs determines the course of the disease.
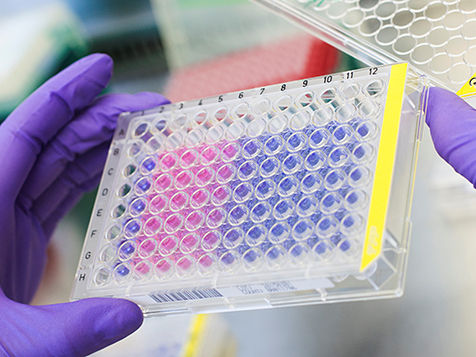
This picture shows the results of a colorimetric assay that measures rates of proliferation of Ewing sarcoma cells.
Judith Glaubitz / LMU
Dr. Thomas Grünewald studies the genetics and molecular pathology of Ewing sarcoma, a malignant bone cancer that is found primarily in children, adolescents and young adults. Most cases of Ewing sarcoma are associated with a single dominant driver mutation which, however, may not be fully sufficient to trigger tumorigenesis. Grünewald leads a Max-Eder Junior Research Group in Pediatric Sarcoma Biology at LMU’s Institute of Pathology. In a paper published in Nature Genetics in 2015, he and his colleagues demonstrated that Ewing sarcoma tumorigenesis is driven by the interplay of an acquired somatic mutation that occurs spontaneously in the tissue concerned and an inherited germline variant that is present in all the patient’s cells.
In a new study, which appears in the online journal Nature Communications, Grünewald’s team shows how such genetic interplay determines the course of the disease in individual patients. “The clinical course of the disease is very heterogeneous,” says Grünewald, “and this variation cannot be attributed to the acquired somatic driver mutation, since this mutation is common to all patients. We therefore took a closer look at the somatic mutation in the context of the individual genome. The acquired mutation occurs in a variety of genetic backgrounds, and it turns out that inherited variations in regulatory elements have a marked effect on how the driver mutation acts in different individuals.
All patients with Ewing sarcoma share the same somatic mutation in the affected tissue, but they differ from one another in terms of the genetic variability they have inherited. The driver mutation results in the fusion of two genes, and leads to the production of a hybrid transcription factor, which binds to, and activates other genes. The new study demonstrates that inherited variation in the regulatory sequences of several of these target genes defines their affinity for the hybrid protein. This factor in turn determines the level of their expression – and the rate of growth of the tumor. This interplay is what accounts for the differences in the severity of the disease from patient to patient. “Up to now, personalized cancer therapy has focused mostly on the acquired somatic mutations. Our study shows that the genetic context is a crucial determinant of disease progression, and that it can inform therapeutic decisions,” says Grünewald. Julian Musa, lead author of the new paper, explains why this is the case: “Depending on their levels of activation, genes that are regulated by such a genetic interplay can be targeted to different degrees by particular drugs. This interplay can at least in part explain why one and the same drug can effectively inhibit the growth of one patient’s tumor, but maybe not that of the patient in the next bed.” The LMU team now plans to investigate this phenomenon in greater detail.
Grünewald and his colleagues also want to explore whether the growth of other tumors is also subject to this sort of dual control. “Our work on Ewing sarcoma is one of the first studies to document the impact of the interplay between germline genetic variations and somatic mutations on the progression of malignant disease in individual patients. We assume that this principle also applies to other types of cancer.”
Original publication



Other news from the department science



























































